Highlights
┬Ę Pituitary apoplexy is often the first presentation of an underlying pituitary tumor.
┬Ę Ischemic stroke can be a rare manifestation of apoplexy of a pituitary macroadenoma in the pediatric population. To the best of our knowledge, our patient is only the third reported case in an adolescent.
┬Ę Prompt recognition and treatment combining medical (cabergoline) and neurosurgical intervention resulted in successful acute and long-term outcomes in our patient.
Introduction
Pituitary tumor apoplexy is an acute clinical emergency due to infarction of the pituitary adenoma with or without hemorrhage [1] and a relatively uncommon presentation of pituitary adenomas that may lead to significant neurological morbidities and even death. The most common clinical manifestations of pituitary apoplexy are sudden onset of headache, nausea, vomiting, deterioration of visual field and acuity, diplopia, ophthalmoplegia, blindness, and altered mental status. Ischemic stroke is an exceptionally rare manifestation of pituitary apoplexy, with only 25 such cases (predominantly in adults) identified in a literature search through 2015 [2]. To the best of our knowledge, the approximate frequency of cerebral infarction in pituitary apoplexy has not been estimated in any published large case series of pituitary apoplexy (even in adults) [1]. Intratumoral hemorrhage can lead to rapid enlargement of the primary mass with compression of intracranial vessels. The mass effect from the lesion may also induce cerebral vasospasm, which can further increase the risk of stroke [3]. To the best of our knowledge, only 2 previous pediatric cases of cerebral infarction associated with pituitary tumor apoplexy have been published [3,4]. Herein, we report a rare presentation of ischemic stroke due to apoplexy of pituitary macroadenoma in an adolescent male. The patient provided informed written consent for publishing the case report.
Case report
A 16.5-year-old male presented to our hospital with acute onset (<4 hours) confusion, visual deterioration, slurred speech, and right-sided weakness. He had been unwell with headache and vomiting for the previous 48 hours. The patient had no associated history of fever, skin rash, seizures, or loss of consciousness. Past medical and family history was unremarkable. Clinical assessment revealed reduced Glasgow Coma Scale (GCS) score (eye opening, 4; verbal response, 2; motor response, 5), right upper and lower limb hypertonia and hemiparesis, and bitemporal hemianopia.
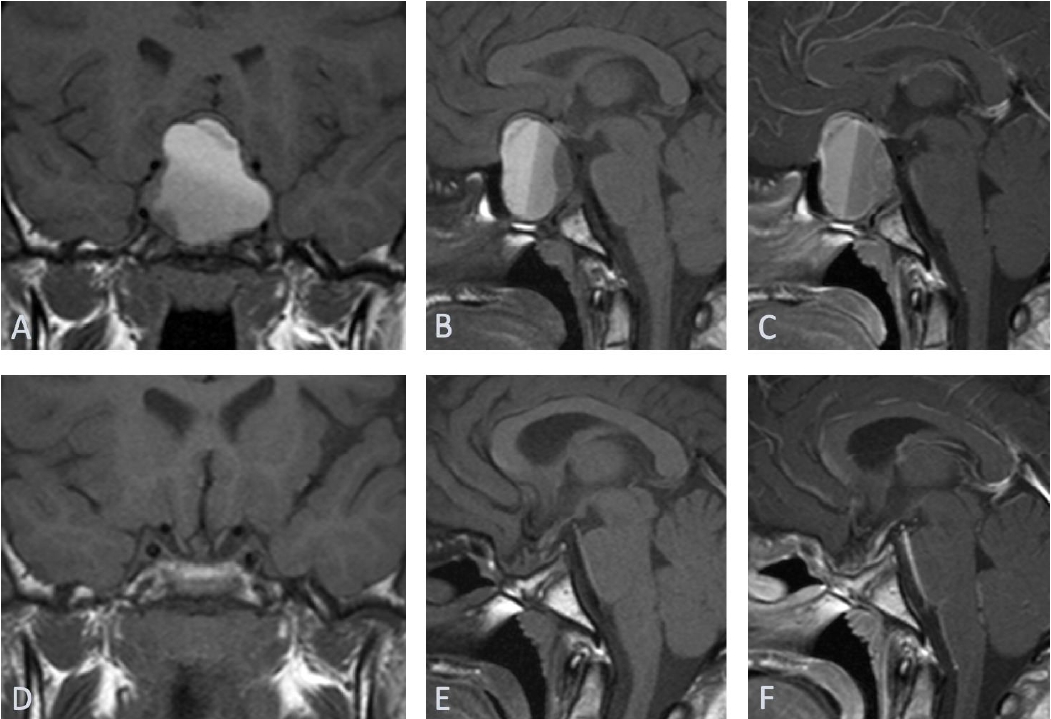
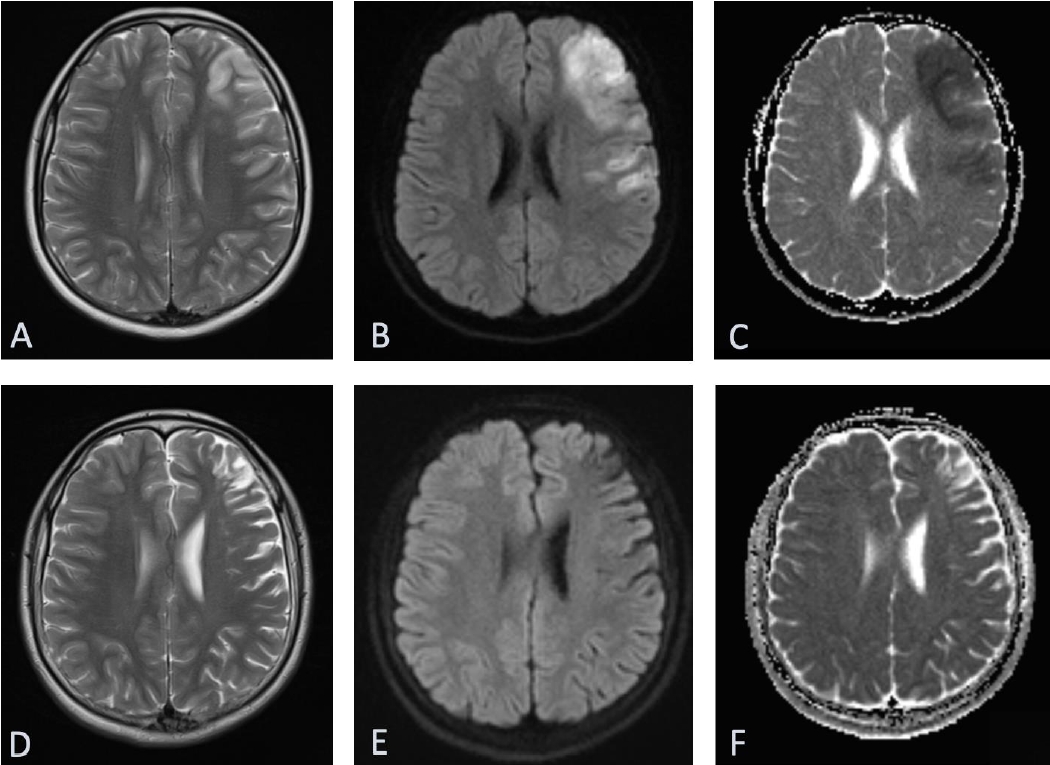
Urgent head computed tomography (CT) scan revealed a 3.5├Ś2-cm sellar/suprasellar mixed density solid and cystic mass with expansion of sella turcica and area of subtle hypodensity in the left frontoparietal and part of left temporal lobe. Subsequent magnetic resonance imaging of the head confirmed a large 2.5├Ś3.5├Ś3.7-cm sellar mass extending substantially into the suprasellar region and markedly elevating the optic chiasm and parasellar regions (Fig. 1). The mass contained largely hemorrhagic contents with fluid levels of slightly different T1 hyper intensities with a more solid enhancing peripheral component. In addition, a fairly demarcated area of cortical T2/FLAIR hyperintensity with corresponding diffusion restriction was observed in the middle cerebral artery (MCA) territory (Fig. 2). CT intracranial angiogram showed luminal occlusion of both internal carotid arteries (ICAs), involving the clinoid and ophthalmic segments due to direct pressure effect from the mass (Fig. 3). Luminal occlusion was more extensive on the left side compared with the right side.
Biochemical investigations (Table 1) showed hyperprolactinemia, low free thyroxine with inappropriately normal thyroid stimulating hormone (TSH), low serum sodium, and serum osmolality. Random serum cortisol was 494 nmol/L. Height at presentation (-1.57 standard deviation score, midparental height not available due to family circumstances), delayed bone age (TWIII 13.9 years), and low serum insulin-like growth factor 1 (IGF-1, 19.1 nmol/L; normal range ,15.6ŌĆō66.9 nmol/L) were consistent with growth hormone (GH) deficiency. The presumptive diagnosis was apoplexy of pituitary macroprolactinoma with hypopituitarism.
The patient was started on stress dose of intravenous hydrocortisone (100 mg every 6 hours) and received a stat dose of oral cabergoline (0.5 mg). Due to decreased GCS score, threat to his vision, and luminal occlusion of bilateral ICA caused by mass effect, urgent endoscopic transsphenoidal tumor debulking was performed within 24 hours. Postoperatively, immediate improvement in GCS score and resolution of visual field defect were observed. Postoperative CT angiogram showed an improvement in the caliber of the distal cavernous, genu. and supraclinioid segments of the left ICA due to reduced pressure effect.
Tumor tissue histology showed abundant hemorrhage and an adenoma composed of sheets of relatively monomorphic cells with eosinophilic cytoplasm. Immunochemistry was strongly positive for prolactin and negative for GH, follicle-stimulating hormone, luteinizing hormone, TSH, adrenocorticotropic hormone (ACTH), and pan alpha subunit. In addition, Ki-67 proliferation index was high (7%).
The patient had a prolonged hospital stay after surgery for neurorehabilitation but completely recovered from motor and cognitive deficits. Postoperatively, hydrocortisone was weaned to a maintenance dose (10ŌĆō15 mg/m2/day), levothyroxine (100 ╬╝g once daily) was started, and cabergoline (0.5 mg twice weekly) was continued. The standard Synacthen test 6 months after initial presentation confirmed persisting ACTH deficiency (Table 1). GH (1 mg subcutaneous daily) was started at 16.8 years of age due to reduced growth velocity (<2 cm/yr) and persistently low serum IGF-1 (16.4 nmol/L; normal range, 15.6ŌĆō 66.9 nmol/L). A formal GH provocation test was not performed. Testosterone replacement was introduced at 17.3 years of age due to lack of spontaneous pubertal progression (genitalia stage III, pubic hair stage III, testes volume 5/6 mL), and the testosterone dose was gradually increased in concordance with doses required to achieve a normal pubertal growth spurt and virilization. An increase in testes volume was not observed, indicating gonadotropin deficiency. Growth velocity notably increased (5.3 cm in the first year and 7.9 cm in the second year) with GH and testosterone replacement. The cabergoline dose was reduced based on serial serum prolactin measurements and eventually was discontinued.
At the last clinical review (18.8 years), the patient was on full anterior pituitary hormone replacement including GH. We plan to assess GH and cortisol status after the patient has finished growing.
Discussion
Pituitary apoplexy is classically defined as abrupt occurrence of acute hemorrhagic or nonhemorrhagic infarction of a pituitary adenoma, which causes characteristic symptoms including acute headache, vomiting, meningism, visual impairment, ophthalmoplegia, or alteration in consciousness. The term pituitary apoplexy should ideally be reserved for classical presentation, whereas subclinical pituitary apoplexy is better suited for asymptomatic pituitary hemorrhage identified at surgery, during radiological investigation, or at the pathological examination [1].
Pituitary apoplexy is often the first presentation of an underlying pituitary tumor, as observed in our patient [5]. Sellar mass on CT scan with presence of euvolemic hyponatremia indicated syndrome of inappropriate antidiuretic hormone and central hypothyroidism, leading to the diagnosis of pituitary apoplexy in our patient. This allowed timely performance of further imaging, corticosteroid replacement, and cabergoline in the presence of hyperprolactinemia.
Ischemic stroke is an extremely uncommon manifestation of pituitary apoplexy in the pediatric population, with few reports in the literature. Due to the rarity of this manifestation, recommending when to suspect pituitary apoplexy as a cause of ischemic stroke is difficult. However, pituitary apoplexy should be considered in the presence of sellar mass and/or pituitary hormone dysfunction in association with ischemic stroke. ICA lumen compression and vasospasm are the suggested mechanisms for cerebral ischemia. To the best of our knowledge, only 2 previous pediatric cases of ischemic stroke from pituitary apoplexy have been reported [3,4]. Another pediatric case of ischemic stroke in the territory of MCA presumed to be caused by direct compression of ICA from a macroprolactinoma unrelated to pituitary apoplexy has also been described [6]. The ICA passes through the cavernous sinus and lies in close vicinity of the pituitary gland. However, the integrity of the ICA lumen is rarely compromised by pituitary adenomas compared with non-pituitary adenoma lesions that encase the ICA [7], explaining the rare occurrence of cerebral ischemia. Extravasation of blood or release of tumor secretions of vasoactive substances in the subarachnoid space during pituitary apoplexy could result in ICA vasospasm causing cerebral ischemia [3].
Debate remains regarding the optimal management strategy for pituitary apoplexy [1]. Neuro-ophthalmic complications (visual fields and visual acuity impairment) and impaired consciousness have been considered indications for decompressive surgery in most adult series [8,9]. In a few retrospective series, similar outcomes in surgically and conservatively managed groups were reported [8]. However, this is likely related to selection bias, with less severe pituitary apoplexy cases treated with conservative management rather than surgery. In addition, attempts have been made to develop a scoring tool (pituitary apoplexy score) to assist in clinical intervention decisions [10]. The tool is based on 3 neuro-ophthalmic parameters (visual acuity, visual field defect, and ocular paresis) and the GCS score that ranges from 0 to 10, with a higher score indicating extensive neuro-ophthalmic impairment. Our patient was managed with transsphenoidal debulking due to severe neuro-ophthalmic manifestations. Among the 2 previous pediatric cases, the one presumed to be due to ICA compression was also managed with surgical decompression, and the case presumedly due to bilateral carotid vasospasm was managed conservatively [3,4]. At the 6-month follow-up, one patient had persistent right-sided hemiparesis, and the other patient had complete resolution of ophthalmoplegia and facial paresis. These types of pediatric cases are extremely rare, and each should be managed (conservative vs. surgical) based on individual medical needs. Similarly, a randomized controlled clinical trial including conservative versus surgical intervention in patients with pituitary apoplexy to determine the optimal management strategy would be difficult if not impossible.
In conclusion, this case report describes an extremely rare presentation of apoplexy of a pituitary macroadenoma in an adolescent, causing an ischemic stroke in the distribution of the MCA. Pituitary apoplexy is a medical emergency and requires prompt recognition and treatment combining medical (cabergoline) and neurosurgical intervention to achieve successful acute and long-term outcomes.