Genetic regulation of linear growth
Article information

Abstract
Linear growth occurs at the growth plate. Therefore, genetic defects that interfere with the normal function of the growth plate can cause linear growth disorders. Many genetic causes of growth disorders have already been identified in humans. However, recent genome-wide approaches have broadened our knowledge of the mechanisms of linear growth, not only providing novel monogenic causes of growth disorders but also revealing single nucleotide polymorphisms in genes that affect height in the general population. The genes identified as causative of linear growth disorders are heterogeneous, playing a role in various growth-regulating mechanisms including those involving the extracellular matrix, intracellular signaling, paracrine signaling, endocrine signaling, and epigenetic regulation. Understanding the underlying genetic defects in linear growth is important for clinicians and researchers in order to provide proper diagnoses, management, and genetic counseling, as well as to develop better treatment approaches for children with growth disorders.
Introduction
Height, as a measure of linear growth, is highly heritable, and genetics plays a major role in the regulation of linear growth [1]. To understand the genetic mechanisms of linear growth, various efforts have been made for decades, and recently genome-wide approaches such as genome-wide association (GWA) studies or exome/whole genome sequencing have been performed successfully, identifying many genomic loci associated with height variation in the general population as well as identifying monogenic changes that cause either syndromic or isolated short stature or overgrowth/tall stature [2-4]. Combining these newly discovered findings obtained from genome-wide approaches into important growth-regulating genes, we now possess a better understanding of human growth disorders [4]. It is now understood that: (1) linear growth occurs at the growth plate and many genes that affect height variation or cause growth disorders play major roles in growth plate biology [5]; (2) the type of mutation can determine the growth outcome; both loss or gain of function mutations can cause either short or tall stature. For example, loss-of-function mutations in NPR2 cause short stature while gain-of-function mutations in the same gene cause tall stature. On the other hand, loss-of-function mutations in FGFR3 cause tall stature while gain-of-function mutations cause short stature [4].; (3) the severity of the genetic abnormality can determine the severity of disease. For example, for some genes, a biallelic mutation can cause severe skeletal dysplasia while a monoallelic mutation in the same gene can cause isolated short stature, as seen in SHOX deficiency or ACAN mutations [6,7].; (4) lastly any genetic defect that alters the biology of chondrocytes in the growth plate can potentially cause growth disorders and these genetic defects can be very heterogeneous [4].
With recent remarkable advances in discoveries of genes responsible for regulating childhood growth disorders, it has become more important for pediatricians, pediatric endocrinologists and geneticists to understand the underlying genetic defects in children who present with growth concerns in order to provide genetic diagnoses, proper management, and genetic counseling. Therefore, this review aims to provide an update on genes known to cause human growth disorders specifically by focusing on those also found through GWA studies to be associated with normal height variation. The genes noted in this review are particularly important because single nucleotide polymorphisms (SNPs) in these genes (presumably causing mild changes) cause height variation in the normal range, but mutations (causing more deleterious changes) in the same gene cause distinctive linear growth disorders. Moreover, combined polymorphisms in these genes could potentially be genetic causes of isolated short stature in an oligo/polygenic manner. The listed genes reviewed here are categorized by their function in the growth plate with the mutations and their corresponding phenotype are described.
Growth regulating mechanisms
Linear growth occurs at the growth plate, a cartilaginous structure between the epiphysis and metaphysis (Fig. 1A). It is composed of 3 distinct layers; resting, proliferative and hypertrophic zones, which are temporally and spatially regulated (Fig. 1B) [8,9]. This regulation results in a unique differentiation process from resting chondrocytes to proliferating chondrocytes and finally hypertrophic chondrocytes. Ultimately, at the terminal zone of hypertrophic chondrocytes, hypertrophic chondrocytes undergo apoptosis and blood vessels invade, resulting in remodeling of the cartilage into newly formed bone [8]. Constant repetition of this process produces new bone and leads to the elongation of bones. Many genes play a role in this process and so far, hundreds of genes have been implicated in either human growth disorders or height variation by affecting chondrogenesis at the growth plate [5].
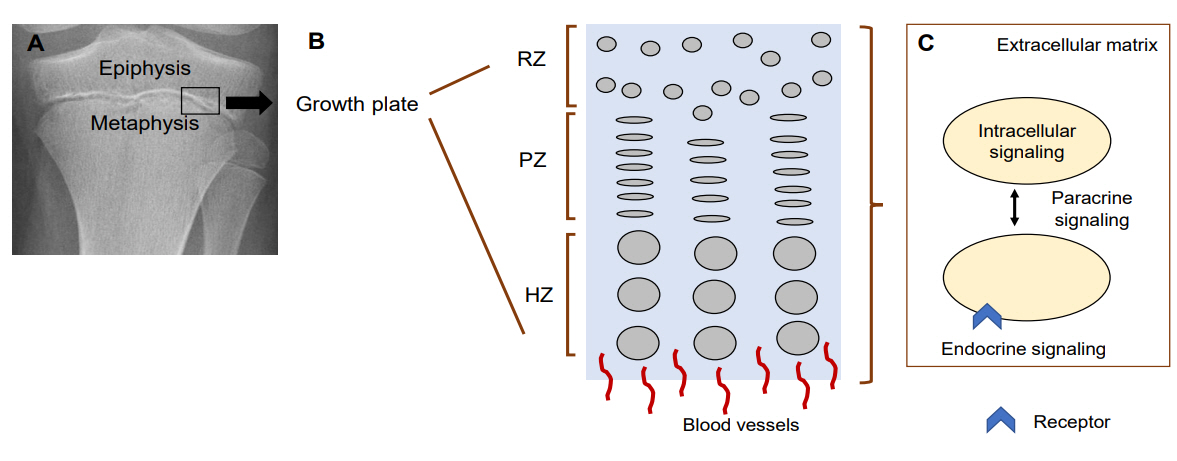
Mechanisms regulating chondrocytes in the growth plate. (A) Growth plate on X-ray. Black box indicates where growth plate is located. (B) Diagram of the 3 layers in the growth plate. (C) Diagram of chondrocyte-regulating mechanisms. RZ, resting zone; PZ, proliferative zone; HZ, hypertrophic zone. Modified from Jee YH, et al. Endocrinol Metab Clin North Am 2017;46:259-81 [15].
The growth mechanisms regulated by these genes can be divided into five different categories: (1) extracellular matrix, (2) intracellular signaling, (3) paracrine signaling, (4) endocrine signaling, and (5) epigenetic regulation (Fig. 1C).
1. Cartilage extracellular matrix
Growth plate chondrocytes secrete an extracellular matrix that is essential for proper functioning of the growth plate. The extracellular matrix consists of collagens, proteoglycans, and glycosaminoglycans and along with secreted growth factors and the modifying enzymes, extracellular matrix serves as a dynamic scaffold that supports and orients growth plate chondrocyte differentiation [10]. Mutations in extracellular matrix-regulating genes can cause isolated short stature or severe skeletal dysplasia, supporting the idea that extracellular matrix plays a critical role in growth plate regulation in humans [11].
1) Aggrecan (ACAN)
Aggrecan, encoded by ACAN, is a chondroitin sulfate proteoglycan, one of the main components of the extracellular matrix in growth plate cartilage that has been shown to modulate hedgehog signaling in a mouse model [12]. In aggrecan-null mice, growth plates were devoid of matrix, showed abnormal chondrocyte organization and differentiation, and the expression of important growth plate regulatory genes such as Col10a1, Sox9, and Ihh was also affected [13]. Therefore, alterations in the structure or function of aggrecan can significantly affect the growth plate cartilage resulting in a change in linear growth. Biallelic mutations in ACAN cause spondyloepimetaphyseal dysplasia and monoallelic mutations cause isolated short stature with or without advanced bone age causing early cessation of linear growth in most cases [6,14]. Patients with mutations in ACAN may also present with premature and severe osteoarthritis and osteochondritis dissecans [15,16]. Heterozygous ACAN mutations are one of common causes of idiopathic short stature; in a cohort of 428 patients with idiopathic short stature, 6 potential disease-causing heterozygous ACAN mutations were found (1.4% of patients) [17]. Thus, in children who have a family history of short stature and early-onset arthritis, ACAN mutations should be considered.
2) Fibrillins (FBN1)
Fibrillins are the major components of microfibrils in the extracellular matrix, and 2 genes in particular, fibrillin 1 (FBN1) and fibrillin 2 (FBN2), have been implicated in growthregulating mechanisms, which regulate transforming growth factor beta (TGF-β) and bone morphogenetic factor (BMP) signaling in the extracellular matrix [3,18]. Monoallelic mutations in FBN1 give rise to acromelic dysplasia, a group of disorders that include Weill-Marchesani syndrome (WMS), acromicric dysplasia and gelophysic dysplasia [18].
Latent transforming growth factor-beta-binding protein 2 (LTBP2) is an extracellular matrix protein, which is a member of superfamily composed of three fibrillins and four other LTBP proteins and also participates in the regulation of TGF-β release in the extracellular matrix. Therefore biallelic mutations in LTBP2 can also cause WMS [19]. Patients with WMS present with short stature with brachydactyly, stiff joints, muscular build, thick skin, ectopia lentis and cardiac valvular problems [18,20]. Interestingly, mutations in FBN1 can also result in Marfan syndrome, an autosomal dominant inherited connective tissue disorder [21]. Individuals with Marfan syndrome have tall stature, skeletal and ocular abnormalities, and over 80% of Marfan syndrome patients have cardiovascular complications [22]. Although it has been proposed that mis-assembled microfibrils leads to decreased extracellular matrix integrity and the misregulation of TGFβ ligands leads to a Marfan syndrome phenotype, the precise mechanisms of how mutations in the same gene could cause 2 opposing phenotypes, short stature versus tall stature, is still unclear [23,24]. Monoallelic mutations in fibrillin 2 (FBN2) have been reported to cause a Marfanlike disorder with tall stature and congenital contractural arachnodactyly, which further confirms the importance of microfibrils in the regulation of linear growth [25]. However, it is not known yet if other mutations in FBN2 can also cause acromelic dysplasia.
3) Metalloproteinases (ADAMTS17)
Metalloproteinases are enzymes that degrade extracellular proteins. There are various kinds of metalloproteinases, such as matrix metalloproteinases (MMPs), a disintegrin and metalloproteinase (ADAMs), a disintegrin and metalloproteinase with thrombospondin motifs (ADALTSs) and ADAMTSlike proteins (ADAMTSLs). MMPs have been shown to play an integral role in regulating chondrocyte proliferation, extracellular matrix degradation, and release of extracellular matrix proteins [26]. However, there have not yet been reports of mutations in MMP-encoding genes causing skeletal defects in humans, although there has been a case report of Winchester syndrome due to a hypomorphic mutation in MMP14 [27]. The precise functions of these enzymes have not been wellestablished but many family members were identified by GWA studies to be associated with height variation (ADAM28, ADAMTS19, ADAMTS2, ADAMTS3, ADAMTS6, ADAMTSL1, ADAMTSL3) suggesting a possible role in linear growth. However, it is also possible that these genes may have been just innocent neighbors to other important genes associated with height variation. Among these, ADAMTS17 has been recognized as a modulator of fibrillin microfibrils, and therefore it is not surprising that biallelic mutations in ADAMTS17 cause Weill-Marchesani-like syndrome, where individuals present with short stature and spherophakia, but do not have brachydactyly and joint stiffness [28-30].
4) Collagens (COL9A2, COL11A1, COL27A1) and matrilin 3 (MATN3)
Cartilage fibrils are complex structures containing collagens II and XI with or without collagen IX and collagen XVI [31]. These, together with other fibrils containing small leucinerich proteins or proteoglycans, such as decorin, biglycan and fibromodulin, make up the fibrillar matrix [11]. The interaction of the fibrillar and extrafibrillar matrix then form a fibrillar periphery [31]. COL11A1 and COL27A1 are important for the organization of the proliferative zone, play a structural role in the pericellular extracellular matrix of the growth plate, and are thought to be important for the transition of cartilage to bone [32-34]. Mutations in these proteins underlie a broad spectrum of skeletal dysplasias (Table 1). Monoallelic mutations in COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3 genes cause Stickler syndrome [35]. Heterozygous mutations in COL11A1 have also been implicated in Marshall syndrome, a chondrodysplasia that is phenotypically very similar to Stickler syndrome. There is some debate as to whether these disorders are separate entities or different expressions of the same disorder. They present with chondrodysplasias characterized by midfacial hypoplasia, myopia, sensorineural-hearing deficits, and short stature in the patient relative to their unaffected family members [36-38]. On the other hand, biallelic mutations cause fibrochondrogenesis 1, a skeletal dysplasia presenting with short limbs, flat midface and protrudent abdomen [39]. Furthermore, biallelic mutations in COL27A1 cause Steel syndrome [40-42]. The main clinical features of this disorder are bilateral hip and radial head dislocations, scoliosis, short stature, carpal coalitions, and pes cavus, with some patients also having facial dysmorphism [42]. Patients with monoallelic mutations in COL9A2 and MATN3 can present with multiple epiphyseal dysplasia, a clinically variable disease characterized by short stature and early onset osteoarthritis [43-45]. Conversely, biallelic mutations in COL9A2 and MATN3 cause a more severe form of skeletal dysplasia [44,47].

Genetic causes of linear growth disorders
2. Intracellular signaling
Various intracellular signaling pathways have been recognized as causative factors of linear growth impairment, and mutations in intracellular signaling have been identified to significantly impair linear growth. However, surprisingly, not many SNPs in these genes were identified to be associated with height variation. It may be because not all the functions of identified genes are well-established or the functions of these genes may be too critical and essential, such as transcriptional regulation, cyclic AMP (cAMP) production, and DNA repair, therefore preventing frequent changes (SNPs) that can affect linear growth.
1) Transcriptional regulation (SOX9)
SOX9 is a critical transcriptional factor for sex development during embryogenesis and for skeletal development and also plays an important role in chondrogenesis at the growth plate by regulating chondrocyte differentiation [48,49]. In humans, dominant mutations in SOX9 cause campomelic dysplasia (which means "bent limb" in Greek) with or without sex reversal [48,49].
2) G protein alpha-subunit (GNAS), cAMP pathway (CREBBP, PDE3A)
The parathyroid hormone-related protein-Indian hedgehog (PTHrP-IHH) negative feedback loop is a major regulator of chondrogenesis in the growth plate, and PTHrP acts through the PTH1 receptor which activates both Gs and Gq family heterotrimeric G proteins [50]. The activation of Gs leads to cAMP production and protein kinase A (PKA) activation, and subsequent phosphorylation of the cAMP response elementbinding (CREB) family of transcription factors [51].
GNAS encodes the G protein alpha-subunit (Gs alpha), which, when activated, increases cAMP production to mediate downstream signaling [52]. Impairment of the cAMP signaling pathway through heterozygous loss-of-function mutations in Gs alpha leads to Albright's hereditary osteodystrophy (AHO) with or without hormone resistance. Maternal inheritance of Gs alpha mutations leads to AHO and pseudohypoparathyroidsim type 1a (PHP1a), which can include multihormone resistance, while patients with paternally inherited mutations of Gs alpha have only the AHO phenotype, termed pseudopseudohypo arathyroidism [52]. Phenotypically, patients with AHO present with short stature, brachydactyly, rounded face, and short neck [53]. Patients who have both AHO and PHP1a also have multihormone resistance to PTH, TSH, and gonadotropins, all of which act through G-protein-coupled receptors [52]. CREBBP encodes a nuclear transcriptional coactivator protein, CREB (cAMP-response element binding protein)-binding protein, that binds specifically to the PKA-phosphorylated form of the CREB protein. Rubinstein-Taybi syndrome (RSTS) is a rare condition characterized by short stature, intellectual disability, broad and deviated thumbs and halluces, and distinct craniofacial characteristics [54]. Over 65% of cases of RSTS are caused by dominant mutations in CREBBP [55].
PDE3A encodes a cyclic GMP (cGMP) and AMP (cAMP) phosphodiesterase 3A. Missense mutations in PDE3A have been shown to cause Mendelian hypertension and brachydactyly type E in seven unrelated families [56,57]. This syndrome has symptoms of short stature, brachydactyly, salt-independent and age-dependent hypertension, an increased fibroblast growth rate, and other cardiovascular complications. The identified mutations increase PKA-mediated PDE3A phosphorylation and increase cAMP-hydrolytic activity, thereby decreasing cAMP production [56].
3. Paracrine signaling pathways
Paracrine factors, such as fibroblast growth factors (FGFs), C-type natriuretic peptide (CNP), IHH, PTHrP encoded by PTHLH, and BMPs, play many critical roles in the growth plate, including chondrocyte proliferation, differentiation, hypertrophy, and matrix assembly [8]. Mutations in these genes have been identified to cause certain growth disorders with unique phenotype.
1) IHH-PTHrP pathway (IHH, PTHLH, PTH1R)
In the growth plate, IHH-PTHrP participates in a negative feedback loop that modulates chondrocyte differentiation [58]. Therefore, mutations in IHH, PTHLH, and the PTHrP receptor, PTH1R, result in disorders that affect skeletal growth. Mutations in IHH lead to brachydactyly type A1 and acrocapitofemoral dysplasia, and also can present with isolated short stature [59-61]. Mutations in PTHLH cause brachydactyly type E with short stature and the most prominent symptoms of these disorders are shortening of the metacarpals or metatarsals and short stature [62]. Loss-of-function mutations in PTH1R cause Blomstrand chondrodysplasia and gain-of-function mutations cause Jansen's metaphyseal chondrodysplasia, both of which are characterized primarily by short-limbed dwarfism [63,64].
2) CNP-NPR2 Pathway (NPPC, NPR2)
The CNP is encoded by the NPPC gene and exerts its effects through the natriuretic peptide receptor B (NPR-B) encoded by NPR2 [65]. Loss of function mutations in NPPC and NPR2 cause short stature, while overexpression in NPPC or gain-of-function mutations in NPR2 cause overgrowth [66-69]. Binding of CNP to NPR-B stimulates guanylyl cyclase activity, increasing the synthesis of cGMP and activating the type 2 cGMP- dependent protein kinase, which then inhibits the MAPK pathway and antagonizes FGFR signaling [70,71]. Because FGFR signaling induces growth arrest of chondrocytes, CNP promotes chondrocyte proliferation. Biallelic NPR2 mutations cause acromesomelic dysplasia, Maroteaux type (AMDM), an autosomal recessive skeletal dysplasia characterized by severe short stature and short limbs [72]. Multiple mutations causing loss of function in NPR-B have been associated with AMDM. Nonsense mutations in the intracellular kinase homology domain (KHD) of NPR-B lead to impaired cGMP production, providing evidence that this domain is essential for skeletal growth [73,74]. Gain-of-function mutations in the NPR2 gene have also been described, and these patients have tall stature and macrodactyly of the big toes [69,74,75]. It has been shown that a gain-of-function mutation in the KHD domain of NPR-B results in an increase in cGMP activity compared to the wildtype protein [76].
3) TGF-β signaling (GDF5)
The GDF5 gene encodes growth differentiation factor 5 (also known as cartilage-derived morphogenic protein) which acts through BMP receptors. Mutations in this gene can cause brachydactyly (type A1, A2, and C) with short stature, chondrodysplasia - Grebe type, characterized by severe dwarfism, disproportionate short stature, and brachydactyly, and Hunter-Thompson type, characterized by skeletal abnormalities that are restricted to the limbs and limb joints [77-81]. Early studies showed that GDF5 is primarily expressed in long bones during human embryonic development [82] and recent studies revealed that GDF5 enhances cartilage formation and GDF5 and TGFβ synergistically induce mesenchymal stem cell differentiation into chondrocytes, providing evidence for its important role in cartilage development [83,84].
4) WNT signaling (WNT5A)
The canonical WNT signaling pathway is important for cell proliferation and cell fate change during development. Studies in Wnt5a-null mice showed a defect in cell polarization in the growth plate [85]. Dominant mutations in WNT5A are associated with Robinow syndrome, a disorder characterized by short stature, limb shortening, genital hypoplasia, and craniofacial abnormalities [86,87].
5) IGF2
IGF2 encodes insulin-like growth factor 2, an important fetal growth factor that binds to the type 1 IGF receptor to regulate cell proliferation. Recently, studies showed that growth plates in Igf2- null mice show a disproportionally larger hypertrophic zone and delayed secondary ossification center, and, in humans, paternally inherited mutations in IGF2 in human cause postnatal growth failure in addition to prenatal growth [88,89]. The dysmorphic features of affected family members were similar to those of individuals with IGF-2 deficiency seen in Silver-Russell syndrome [89]. IGFBP2BP2 and IGF2BP3, encoding the IGF2 binding proteins, are identified to be associated with height variation but mutations have not yet been found in human disorders.
4. Endocrine signaling
Endocrine signaling pathways are the clinical causes most extensively evaluated in children who present with short stature since endocrine causes and hormonal abnormalities are relatively easily tested in clinical practice. Growth hormone, thyroid hormone, glucocorticoids, and sex steroids affect chondrocyte proliferation and hypertrophy in the growth plate, altering linear growth if there is a defect in the corresponding axis.
1) GH-IGF1 axis (IGF1R)
The GH-IGF1 axis is one of the main regulatory systems that controls chondrogenesis in the growth plate, whereby GH acts directly on the growth plate to stimulate chondrogenesis or indirectly through circulating and local insulin-like growth factor-1 (IGF-1) [90,91]. There have been multiple genes identified that cause growth hormone deficiency or impair downstream signaling of the GH-IGF1 axis. For the scope of this review, only genes identified by GWA studies are mentioned. Mutations in the IGF-1 receptor gene, IGF1R, can cause both prenatal and postnatal growth failure. Monoallelic mutations may cause short stature in children born small for gestational age [92]. Biallelic mutations cause severe short stature [93]. Fibroblast cultures from these patients showed decreased IGF1R function and a reduced number of IGF1 receptors, which could lead to IGF1 insensitivity [93]. SNPs in IGFBP3, IGFBP4, STC2, PAPPA, PAPPA2, which regulate the free form of IGF-1 by functioning as a binding protein (IGFBP3, IGFBP4), binding-protein-cleaving enzymes (PAPPA, PAPPA2), or as a regulator of PAPPA (STC2), have been identified by GWA studies to be associated with height variation, likely by affecting IGF-1 bioavailability and therefore the GH-IGF1 axis. However, only mutations in PAPPA2, encoding Pappalysin-2, a protein that cleaves IGFBP-3 and -5, have been identified in humans where the patients have decreased free IGF-1 levels [94].
2) Insulin signaling (PIK3R1)
PIK3R1 is a gene involved in insulin signaling. Patients with mutations in PIK3R1 have SHORT syndrome, hallmarks of which include short stature, hyperextensibility of joints, ocular depression, Rieger anomaly, teething decay, and insulin resistance [95-97]. However, the role of PIK3R1 in the growth plate is not yet known.
3) Aromatase (CYP19A1)
Estrogen plays an important role in skeletal maturation and mineralization, and defects in estrogen signaling can lead to a variety of disorders that manifest with short or tall stature by altering the timing of growth plate senescence [98]. Since estrogen normally accelerates growth plate senescence and skeletal maturation, children with aromatase deficiency, a rare autosomal recessive disorder caused by mutations in the CYP19A1 gene, present with delayed bone age and delayed puberty which lead to tall stature. This presentation is due to a lack of estrogen converted from androgen [99,100]. On the other hand, overexpression of CYP19A1 can result in aromatase excess syndrome. Affected adults present with short stature, gynecomastia in males, and macromastia in females [101]. Patients with mutations in the estrogen receptor gene ESR1 have estrogen resistance. To date, there have been 5 patients described who have mutations in ESR1, with varying phenotypes and varying degrees of estrogen insensitivity, likely due to different mutation sites and differences in penetrance of the particular mutation [102,103]. However, all patients had delayed skeletal maturation, with some having tall stature in adulthood also due to a lack of estrogen effects on the growth plate [104].
5. Epigenetic control of height (DNMT3A , HIST1H1E , EZH2 , NSD1 , SETD2)
DNA and histone methylation are one of the main gene regulatory mechanisms and play crucial roles in growth and development. The best example of epigenetic regulation of growth is the epimutations in the telomeric imprinting control region 1 (ICR1), which alters H19 and IGF2 expression. Hypermethylation of the ICR1 causes biallelic expression of IGF2 presenting as Beckwith-Wiedemann syndrome (BWS) (overgrowth) while hypomethylation of the ICR1 causes suppression of IGF2 expression presenting as Silver-Russell syndrome (short stature) [105]. Moreover, many genes involved in DNA methylation have been identified to cause growth disorders, especially overgrowth syndromes. Studies have found de novo mutations in DNMT3A, a gene that encodes a DNA methyltransferase, which establishes methylation during embryogenesis [106]. These mutations cause an overgrowth syndrome called DNMT3A syndrome. Patients present with tall stature, intellectual disability, and distinctive facial appearance with a round face, heavy, horizontal eyebrows, and narrow palpebral fissures [106,107]. In addition to DNA methylation, histone methylation has been implicated in growth disorders. HIST1H1E encodes histone H1.4, and in humans, H1.4 mediates the formation of higher-order chromatin structures, thus regulating the accessibility of histone modification factors, regulatory proteins, and chromatin modifiers to target sites on this chromatin structure [108]. Mutations in this gene cause Rahman syndrome. Individuals with this disease have overgrowth with varying degrees of intellectual disability and a distinctive facial appearance in childhood - full cheeks, high hairlines, and telecanthus [108]. Additionally, monoallelic mutations in genes that encode other histone methyltransferases cause Weaver syndrome and Sotos syndrome. Weaver syndrome is caused by mutations in EZH2 (Enhancer of Zeste homolog 2) [109,110]. This gene encodes a histone methyltransferase that is part of the polycomb repressive complex 2, which catalyzes the methylation of lysine residue 27 of histone 3 (H3K27), thus causing transcriptional repression [111]. Patients with Weaver syndrome have tall stature, advanced bone age, dysmorphic facial features, and variable learning disability [109,110]. Sotos syndrome is another overgrowth syndrome characterized by overgrowth, advanced bone age, dysmorphic facial features, and intellectual disability, and patients with Sotos syndrome also often have brain anomalies and seizures [112]. However, in contrast to Weaver syndrome, Sotos syndrome and Sotoslike syndrome are caused by mutations in genes that cause transcriptional activation: NSD1 and SETD2 [112,113]. These 2 genes encode histone methyltransferases which catalyze the methylation of histone 3 lysine 36 to allow for transcriptional activation [112,113]. Interestingly, mutations in NSD1 have been detected in 2 patients with BWS, another overgrowth condition characterized by macroglossia, abdominal wall defects, visceromegaly, embryonic tumors, hemihyperplasia, and neonatal hypoglycemia [114].
Conclusion
In this review, we reviewed the genes that cause linear growth disorders in humans, focusing on their functions at the growth plate and their clinical phenotype. These genes are also identified by GWA studies which suggest that a mild change of function or expression in these genes may modulate linear growth within the normal ranges. The genetic evidence supports the idea that genes causing linear growth are highly heterogeneous, so that any regulatory mechanisms that alter chondrogenesis in the growth plate could be the genetic cause of growth disorders, either syndromic or isolated. With the aid of genome-wide approaches, it is expected that additional regulating mechanisms in growth disorders will be discovered and broaden our knowledge of the underlying causes of these disorders.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH (ZIA HD000640).