Introduction
Single gene mutations and submicroscopic chromosomal rearrangements account for a certain percentage of the etiology of pediatric endocrine disorders1,2,3). Identification of such molecular defects is beneficial for patients, because molecular diagnosis often allows better clinical management and accurate genetic counseling4,5). Furthermore, identification of the genetic causes of congenital endocrine disorders provides advances in medical research.
Recently, various molecular technologies have been developed to identify genetic and genomic alterations in humans. These technologies include next generation sequencing (NGS) that can detect nucleotide substitutions and small insertions/deletions (indels)6,7,8), and array-based comparative genomic hybridization (array CGH) that identifies copy-number variations (CNVs) in the genome9,10,11,12). NGS and array CGH enable us to perform systematic molecular analyses for multiple clinical samples. This review introduces some examples of the use of NGS and array CGH for the molecular diagnosis of pediatric endocrine disorders.
Molecular diagnosis of pediatric endocrine disorders using NGS
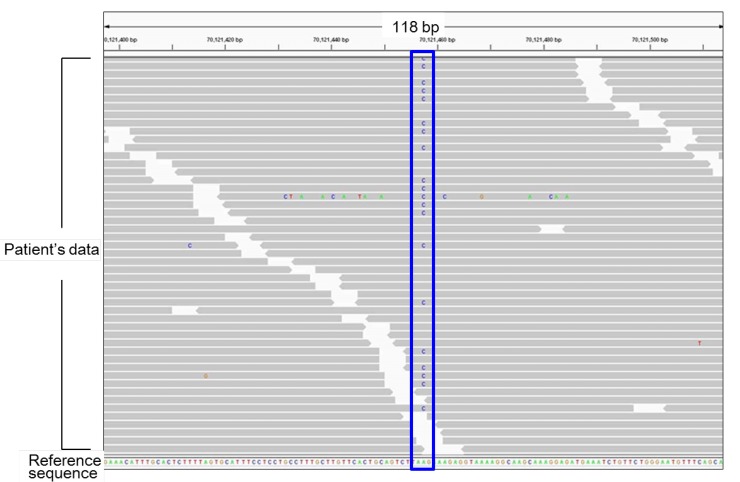
NGS is a powerful tool for high-throughput mutation analysis6,7,8). This method is particularly useful to elucidate the genetic causes of Mendelian disorders with multiple causative genes. In our recent studies13,14,15,16), we performed NGS analyses for several samples of patients with pediatric endocrine disorders (Fig. 1). For example, we investigated the genetic causes of hypogonadotropic hypogonadism (HH), a rare condition caused by monoallelic, biallelic, or oligogenic mutations in more than 20 genes13,16). We created a custom-made NGS panel that covers most known causative genes for HH. DNA samples obtained from 58 patients with HH with and without additional clinical features were analyzed13). As a result, possible pathogenic mutations were identified in 13 of the 58 patients. Mutations in FGFR1 and CHD7 accounted for the majority of the molecular defects in our patients13). We identified an SOX3 polyalanine deletion, a rare mutation known to cause pituitary dysfunction17), in a patient with normosmic isolated HH (IHH)13). This finding expanded the phenotypic spectrum of SOX3 polyalanine deletion to include HH without other pituitary hormone deficiency. Likewise, we detected a WDR11 splice-site mutation in a patient with combined pituitary hormone deficiency (CPHD)13). These data indicated that haploinsufficiency of WDR11, a gene that has recently been implicated in normosmic IHH and Kallmann syndrome (HH with anosmia)18), can also underlie CPHD.
In another study, we analyzed 62 patients with nonsyndromic hypospadias (hypospadias without additional clinical features), a common anomaly in boys15). We created an NGS panel covering the coding exons of 25 known disease-associated genes, and detected mutations in AR, NR5A1, BNC2 and other genes in seven cases15). These data implied that more than 10% of cases with nonsyndromic hypospadias arise from mutations in known causative genes. Importantly, one patient carried probably damaging mutations in multiple genes15), suggesting the possible oligogenicity of nonsyndromic hypospadias.
The abovementioned results provided evidence that mutation screening using NGS is helpful to clarify the contribution of each causative gene to the etiology of polygenic disorders. In addition, NGS can provide critical information about phenotypic variations of each monogenic abnormality
Identification of novel genetic causes of pediatric endocrine disorders by NGS
NGS can also be used for systematic mutation screening of all known protein-coding genes (whole exome analysis)8,19). To date, whole exome analysis has successfully detected several novel causative genes for human disorders. For example, in 2013, Abreu et al.20) performed whole exome analysis of 15 patients with central precocious puberty (CPP) of unknown etiology. NGS analysis of the patients and their unaffected family members identified MKRN3 mutations cosegregated with the disease. Subsequent studies confirmed that MKRN3 mutations constitute one of the major genetic causes of CPP21,22,23).
Similarly, whole exome analysis helped to identify a novel cause of disorders of sex development (DSD). Bashamboo et al., Baetens et al., and our group revealed that an identical missense substitution in NR5A1, p.R92W, induces testicular development in genetic females24,25,26). This finding provided the first indication that specific missense mutations in a single gene can underlie 46,XX testicular/ovotesticular DSD. Since the p.R92W mutation in Nr5a1 did not induce testicular formation in female mice27), it appears that NR5A1 plays a species-specific role in sexual development.
Array CGH-based copy-number analysis for molecular diagnosis of pediatric endocrine disorders
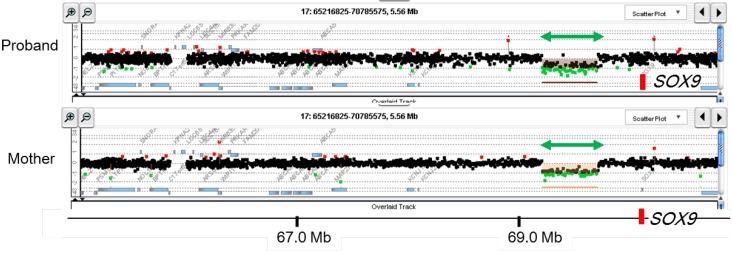
Array CGH can screen CNVs in the entire genome in a single assay9,10,11,12). This method is currently used as the standard procedure for molecular cytogenetic analysis. Indeed, array CGH has higher resolution than conventional G-banding12), although array CGH cannot detect copy-number neutral translocations and inversions. To date, it is known that CNVs account for a significant fraction of the etiology of Mendelian disorders12,28). We have performed array CGH for several patients with pediatric endocrine disorders14,29). We analyzed DNA samples obtained from 24 patients with 46,XY DSD of unknown etiology, and identified submicroscopic deletions in 3 patients29). These results suggested that submicroscopic CNVs represent important genetic causes of 46,XY DSD. These deletions were predicted to induce undermasculinized external genitalia by eliminating exons or cis-regulatory elements of genes involved in sex development. Notably, one patient with complete 46,XY gonadal dysgenesis was found to carry a microdeletion in a far upstream region of SOX9 (Fig. 2)14), indicating that a testis-specific enhancer(s) of SOX9 is located within this deleted region.
Similarly, we utilized array CGH to characterize CNVs in the short arm pseudoautosomal region of sex chromosomes (PAR1) associated with idiopathic short stature or Leri-Weill dyschondrosteosis (short stature accompanied by Madelung deformity in the forearm)30,31,32). It is known that CNVs in PAR1 encompassing SHOX and/or its putative enhancers are important causes of short stature31,33,34). These CNVs were predicted to affect skeletal growth by reducing SHOX expression levels in chondrocytes30,31). We found that CNVs in PAR1 are highly variable in their sizes and positions30,31,32). Breakpoint characterization of the CNVs suggested that various mechanisms such as non-allelic homologous recombination and non-homologous end joining, and possibly replication errors as well, are involved in these abnormalities30,31,34,35). Thus, SHOX abnormality represents a model of human genomic disorders.
Identification of novel genetic causes of pediatric endocrine disorders by array CGH
Array CGH has also identified novel genomic abnormalities associated with pediatric endocrine disorders. For example, we performed array CGH analysis for patients with hereditary gynecomastia (aromatase excess syndrome), a rare autosomal dominant disorder characterized by estrogen overproduction due to excessive enzymatic activity of aromatase36,37,38,39). We found that these patients have submicroscopic CNVs in the upstream region of the aromatase gene (CYP19A1)36,37,38,39). These CNVs included microduplications encompassing CYP19A1 promoters and microdeletions that lead to aberrant fusion between the coding exons of CYP19A1 and promoters of widely expressed neighboring genes38). These data suggested that hereditary gynecomastia is caused by submicroscopic genomic rearrangements leading to CYP19A1 overexpression38). Most importantly, our results indicated for the first time that a tandem duplication involving physiological promoters can underlie gene overexpression and resultant congenital disorders36,38).
Ethical issues associated with the use of NGS and array CGH
Although the use of NGS and array CGH is highly beneficial for patients with pediatric endocrine disorders as well as other genetic diseases, these new technologies also raise ethical issues that are not associated with conventional molecular analyses. For example, whole exome analysis can detect mutations that are not related with the patient's present phenotype40,41). Some of these unexpected genetic alterations may have a potential to cause some serious disorders later in life. In addition, NGS and array CGH frequently detect molecular alterations of unknown pathogenicity 5). There is debate as to which and how the findings of systematic molecular analyses should be reported to the patients. The American College of Medical Genetics and Genomics and other organizations have published recommendations for the management of data obtained from NGS analysis41,42,43).
In addition, NGS and array CGH can be applied to prenatal diagnosis44,45). For example, NGS is used for noninvasive prenatal testing for chromosomal aneuploidy45). NGS and array CGH have broad applicability in the prenatal molecular diagnosis of both Mendelian disorders and multifactorial conditions5). However, the benefits, limitations, and ethical issues of these technologies need to be discussed.
Future implications
NGS and array CGH are beneficial both for clinical practice and for research on pediatric endocrinology. In particular, since the cost of NGS is continuously decreasing, the utility of this method will be further enhanced in the future. Moreover, NGS has additional applications such as characterization of gut microbial flora in individuals with specific conditions46,47). The use of NGS and array CGH will provide further information about the causative mechanisms of pediatric endocrine disorders.