Introduction
Normal linear growth in children along the percentile of their genetic potential is highly complex process, regulated by the interaction of genetic, nutritional, environmental and hormonal factors1). Various hormones, including growth hormone (GH), thyroid hormone, and gonadal steroids, affect growth, with GH playing a key role in the growth phase2). GH, a peptide hormone synthesized and secreted from anterior pituitary, stimulates growth by promoting the production of insulin-like growth factor (IGF)-I in peripheral tissues such as liver and cartilage tissues. IGF-I stimulates the division and expansion of cartilage tissues and performs metabolic activities, including glucose production, protein anabolism, fat metabolism, and bone metabolism. GH deficiency (GHD) is caused by either congenital causes, such as idiopathic, genetic, and developmental anomalies, or by acquired causes, such as head trauma, pituitary tumors, or hypothalamic tumors. Pituitary stalk interruption syndrome (PSIS), a congenital developmental anomaly of the pituitary gland, is a distinct developmental defect characterized by a thin or absent pituitary stalk, hypoplasia of the adenohypophysis, and ectopic location of the neurohypophysis. It manifested as isolated GHD or combined pituitary hormone deficiency (CPHD).
The most frequently found hormone deficiency in PSIS patients is GHD, and short stature resulting from delayed growth is its typical clinical manifestation. However, although it is rare, growth without GH, in which patients maintain normal linear growth despite abnormal GH secretion, is also observed. This phenomenon is observed in some patients who have undergone brain surgery to remove tumors found mainly in the hypothalamic-pituitary area, such as craniopharyngioma. It is very rare to find cases of growth without GH in patients who have not undergone brain surgery although such cases have been reported in individuals with septo-optic dysplasia (SOD) and empty sella syndrome.
We report a case in which a PSIS-induced CPHD patient attained her final height above mid parental height despite a severe GHD.
Case report
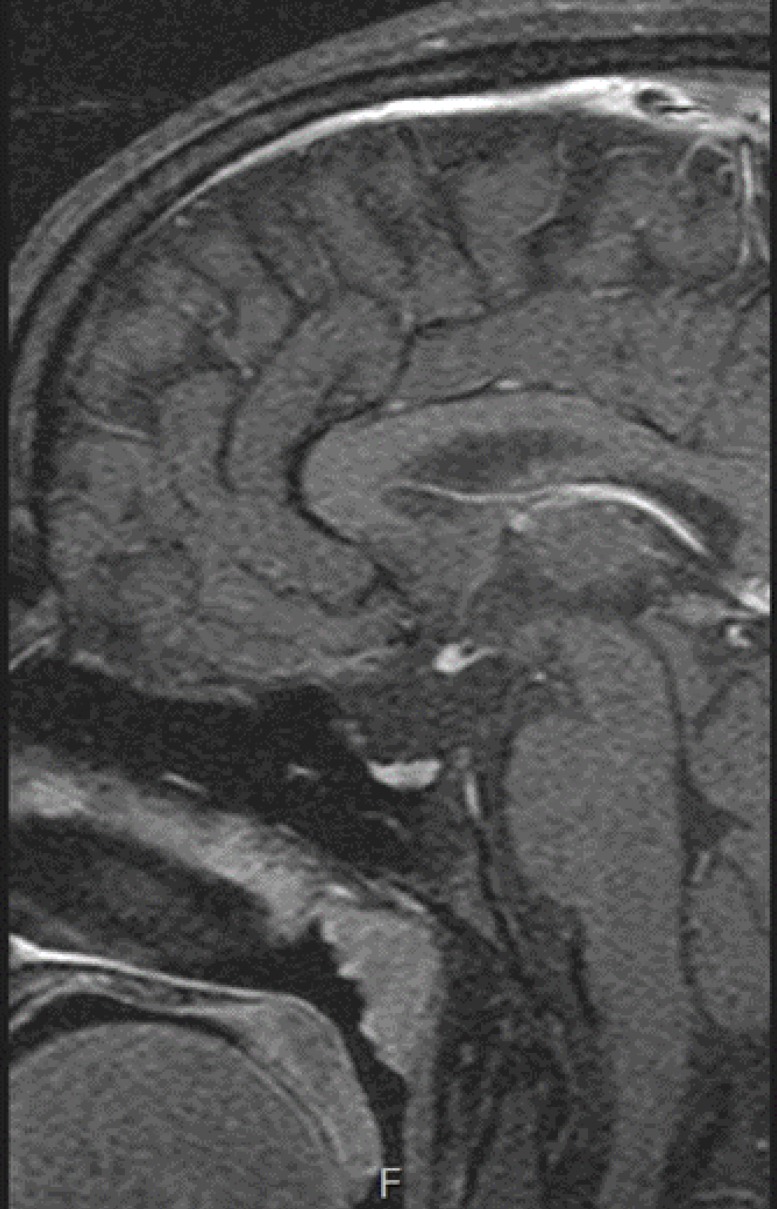
A female patient aged 13 visited Bucheon St Mary's Hospital Emergency Department for seizures. She showed symptoms of fever (40Ōäā), vomiting, and hypoglycemia, with a serum glucose level of 58 mg/dL. We conducted a brain magnetic resonance imaging (MRI) to obtain a differential diagnosis of abnormalities in the central nervous system. The MRI results showed ectopic neurohypophysis and an invisible pituitary stalk, which were consistent with PSIS (Fig. 1). We transferred her to the Department of Pediatric Endocrinology to test pituitary functions.
The patient was born at our hospital by cesarean section at 42 weeks and 1 day of gestation, with a birth weight of 3.56 kg (10thŌĆō50th percentile). She showed signs of lethargy 2 days after birth, and was found to have hypoglycemia on blood sugar test (21 mg/dL). The patient was thus admitted to the neonatal intensive care unit for 14 days. She underwent a transfusion therapy, and her blood sugar stabilized before discharge. During her hospital stay, only a thyroid function test was conducted for endocrinologic evaluation, the results of which were within the normal range. Follow-up observation did not continue afterward, and medical records for the first 13 years of her life could not be obtained. According to her parents, the patient received Emergency Treatment for febrile seizures and hypoglycemia many times at other hospitals until the age of 6. Her motor and language development was normal although her school academic performance was poor. No particular disease was of note in the patient's family history, and she had a younger brother. Her father's height was 170 cm; her mother's height was 160 cm; her midparental height was 158.5 cm.
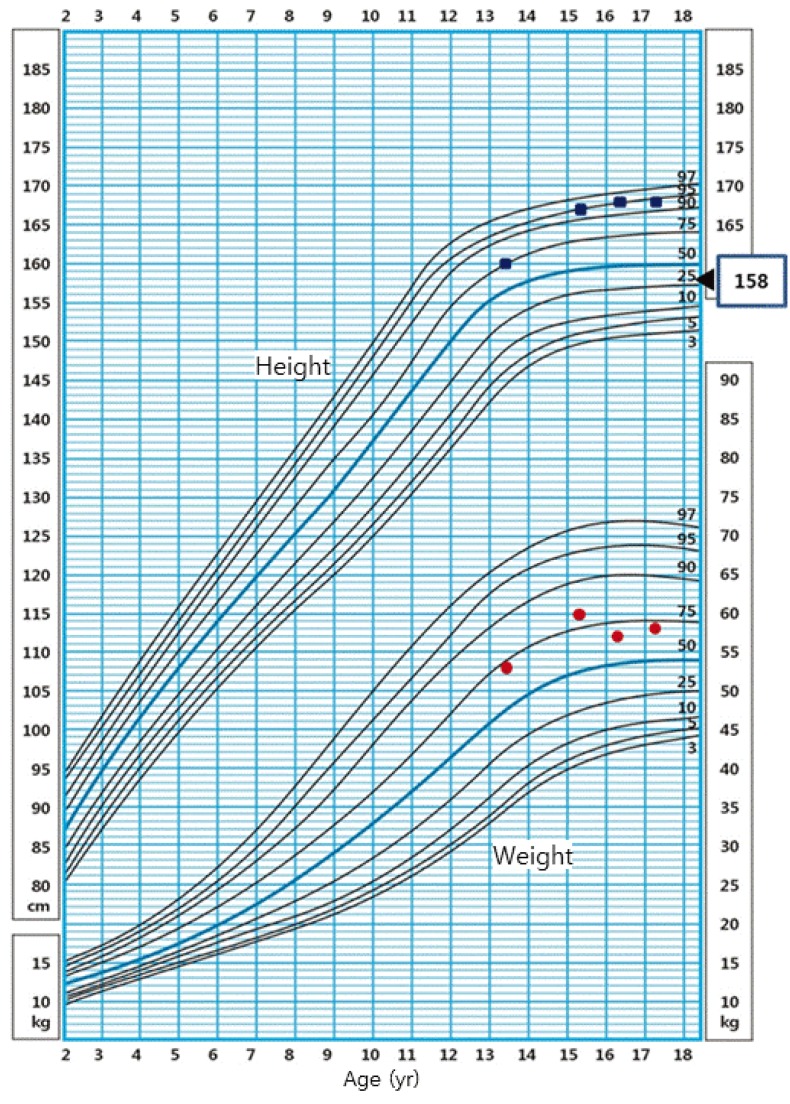
On physical examination, the patient's height was 160 cm (75thŌĆō90th percentile), and she weighed 53 kg (50thŌĆō75th percentile), with a body mass index (BMI) of 20.45 kg/m2 (50thŌĆō75th percentile) (Fig. 2). The sexual maturity rate (SMR) was Tanner breast I and pubic hair I. Radiography of the left hand revealed a bone age of 12 years. Her routine laboratory evaluation showed total cholesterol level of 207.1 mg/dL (normal range for age, <170 mg/dL), increased low-density lipoprotein (LDL)-cholesterol level of 155 mg/dL (normal range for age, <110 mg/dL), increased triglyceride of 156 mg/dL (normal range for age, <90 mg/dL), and decreased high-density lipoprotein (HDL)-cholesterol level of 38 mg/dL (normal range for age, >45 mg/dL) (Table 1).
The patient underwent basal and dynamic endocrine investigation (Table 2). The results showed inadequate GH and cortisol response to insulin-induced hypoglycemia (peak GH, 0.08 ┬Ąg/L [normal limit, >10 ┬Ąg/L]; peak cortisol, 3.28 ┬Ąg/dL [normal limit, >22 ┬Ąg/dL]) consistent with the diagnosis GHD and adrenocortical insufficiency. IGF-I and IGFBP-3 were also decreased at 5.31 ng/mL (normal range for age, 247ŌĆō396 ng/mL) and 1,167.87 ng/mL (normal range for age, 1,527ŌĆō4,418 ng/mL), respectively. LH and FSH levels rose to 1.58 IU/L and 1.16 IU/L, respectively, following stimulation with GnRH. These findings were consistent with hypogonadotropic hypogonadism. TSH showed no response to TRH; regarding prolactin, the basal level was elevated at 64 ng/mL (normal level, 3ŌĆō24 ng/mL) and the level increased further to 157 ng/mL upon TRH simulation. Based on these results, we diagnosed the patient as CPHD caused by PSIS, prescribed cortisol, thyroid hormone, and estrogen, and followed her up in our outpatient clinic.
However, the patient showed poor compliance, and appropriate treatment could not be continued. She visited again as an outpatient 2 years later at the age of 15 years. For 2 years, she grew in height by 7 cm (167 cm; 95thŌĆō97th percentile) and in weight by 7 kg (60 kg; 75thŌĆō90th percentile). The patient's BMI was 21.3 kg/m2 (50thŌĆō75th percentile), and the patient had truncal obesity. SMR was Tanner breast I and pubic hair I and her bone age was 14. CPHD was also confirmed on the combined pituitary function test conducted again. In addition, a standard 75 g oral glucose tolerance test revealed normal glucose tolerance and normal insulin level. Bone density showed osteoporosis (T-score of -3.3). Similar to the prior examination results, there were increases in total cholesterol and LDL-cholesterol, and a decrease in HDL-cholesterol.
The patient started the cortisol (hydrocortisone 10 mg AM and 5 mg PM), thyroid hormone (levothyroxine, 50 ┬Ąg/day), and estrogen (conjugated estrogen, 0.625 mg/day) replacement therapy again. Patient and parent education was provided to increase her compliance to treatment. The patient reached her final adult height (168 cm) at the age of 16, which was 9.5 cm taller than the midparental height, 158.5 cm (Fig. 2).
Discussion
We have described the growth pattern of the female patient with CPHD and PSIS who maintained GH-independent linear growth from diagnosis to final height (Fig. 2). Her presenting features were recurrent hypoglycemic seizure and delayed puberty and no evidence of growth failure. Although she had persistent GHD with marked reduced GH and IGF-I levels on repeated provocative test, she attained a normal final height above her mid parental height. Our patient was diagnosed as CPHD with PSIS at age 13. However, the documented hypoglycemia during neonatal period suggests that GHD was present at that time.
GH is known as an essential element for normal growth. However, cases of normal growth without GH have been reported. Geffner defined these cases as ŌĆ£growth without GH syndrome.ŌĆØ3) Matson4) reported normal growth without GH for the first time, observing that some patients who had CPHD after craniopharyngioma surgery maintained normal growth velocity. Similar reports on other suprasellar tumor surgeries5) have followed. Normal growth without GH was also reported in SOD6), empty sellar syndrome7), and PSIS8).
The mechanism of growth without GH remains still unclear. However, regarding normal growth without GH arising after brain surgery, hypothalamic damage caused by the brain surgery is thought to induce obesity, and the resulting hyperinsulinemia and/or hyperprolactinemia are assumed to be important mechanisms of normal growth without GH in postsurgery patients. It is assumed that elevated insulin and/or prolactin concentration normalize IGF-1 levels even in GHD patients, restricts the increase in serum levels of IGFBP-3, and changes the distribution of circulating IGF-1. Thus, normal growth without GH continues because of the effects of IFG-I, which are independent of GH3,9). In addition, the elevated leptin levels secreted from the increased fat tissues, which result from obesity, have also been suggested as a mechanism of maintenance of normal growth velocity. Holloway10) observed that leptin directly induces the proliferation and maturation of chondrocytes, and that it stimulates IGF-I and IGF-I receptor gene expression in a laboratory model of endochondral ossification; based on these findings, Maor explained that leptin can play a role in maintaining growth velocity by working as a bone growth factor in GHD patients. Our patient was not obese, and her insulin level was within the normal range, yet her prolactin level was elevated. Prolactin levels can increase when the hypothalamus is damaged owing to its defective regulation, and the levels can also increase in hypothalamic tumors and PSIS. Hyperprolactinemia is also suggested as a mechanism of growth without GH; however, concrete details on how it stimulates growth are not known.
Growth without GH has also been reported in SOD patients with normal insulin and prolactin levels and decreased IGF-I levels. Thus, their normal growth seems to be maintained by another mechanism different than that in patients who previously underwent brain tumor surgery. Although uncertain, a report suggested that decreased IGFBP levels may maintain normal free IGF-I concentration despite low IGF-I levels, thus enabling normal growth11).
A few cases of normal growth without GH in patients with PSIS-induced CPHD, as in our patient, have been reported8). These patients, despite their marked reduced GH and IGF-I levels, were not obese, and their prolactin and insulin levels were within the normal ranges. Free IGF-I was also within the normal range. Murashita et al.8) stimulated erythroid progenitor cells with the serum of PSIS patients who showed near-normal growth despite CPHD, and reported that the experimental group showed a normal degree of serum growth-promoting activity when compared to the control group. However, the exact mechanism was not explained clearly.
Arroyo et al.12) reported on growth without GH in a patient with CPHD and hypoplastic pituitary due to a mutation in the PROP1 gene. This patient, whose height was below normal until 15, kept growing until 20 to reach the range of mid parental height. The authors assumed that growth within the mid parental height range was possible due to delayed epiphyseal fusion caused by estrogen deficiency and the resulting prolonged growth. Our patient also had hypogonadotropic hypogonadism; however, she already reached the mid parental height range at the age of 13 and kept growing since to reach the final adult height beyond the mid parental height. Therefore, we assume that factors other than epiphyseal fusion delay are involved.
Apart from the mitogenic activity promoting linear growth, GH exerts a number of other metabolic effects directly or indirectly through IGF-I medication, including effects on body composition, skeletal maturation and mineral metabolism13,14). Pavlou et al.13) reported that a boy aged 7 showed higher than normal growth velocity (8ŌĆō9 cm per year) despite a severe GHD arising after a surgery for suprasellar craniopharyngioma; however, the patient showed metabolic GHD symptoms, such as bone age delay and abdominal obesity. Our patient was in a similar case in that she showed metabolic GHD symptoms, such as bone age delay, central obesity, osteoporosis, and dyslipidemia, although she reached normal height above the mid parental height without GH.
In summary, we report a case in which a CPHD with PSIS patient maintained normal growth, despite severe GH and IGF-I deficiency and metabolic GHD symptoms. The exact mechanism of growth without GH needs to be clarified further in the future. Additional studies are required to determine if children with growth without GH should receive GH treatment as adults do for GHD to obtain GH's metabolic effect.