Highlights
· The main source of endogenous pediatric Cushing syndrome is the pituitary.
· For the diagnosis of Cushing disease (CD), endogenous hypercortisolism must be established, and adrenocorticotropic hormone dependence determined.
· The main treatment for CD is surgery, with the goal of normalizing cortisol levels.
Introduction
Cushing syndrome (CS) is an endocrine disorder that occurs due to prolonged exposure to excess glucocorticoids and can be acquired exogenously or endogenously [1,2]. The overall incidence of endogenous CS is 0.7–2.4 per million people per year, and only 10% of new cases occur in children each year [1]. Depending on its origin, it can be dependent or independent of adrenocorticotropic hormone (ACTH) [3,4].
Cushing disease (CD), which is caused by an ACTH-producing pituitary adenoma, is responsible for 75%–80% of cases of endogenous CS [5] and is more common in children older than 5 years and more frequent in boys, with a prevalence of 63% [6,7]. Its rarity in childhood and adolescence (annual incidence of 0.89–1 per million pediatric patients) [8] has led pediatricians and pediatric endocrinologists to have limited experience in its diagnosis and treatment [3,9].
Growth and development are transcendental processes during childhood. Since hypercortisolism can negatively affect both, its timely diagnosis and treatment are crucial to improve the prognosis [10].
Here, we present an updated narrative review of the pathophysiology, diagnosis, and treatment of CD in the pediatric population. The appropriateness of this review article was assessed using the SANRA scale [11].
Pathophysiology
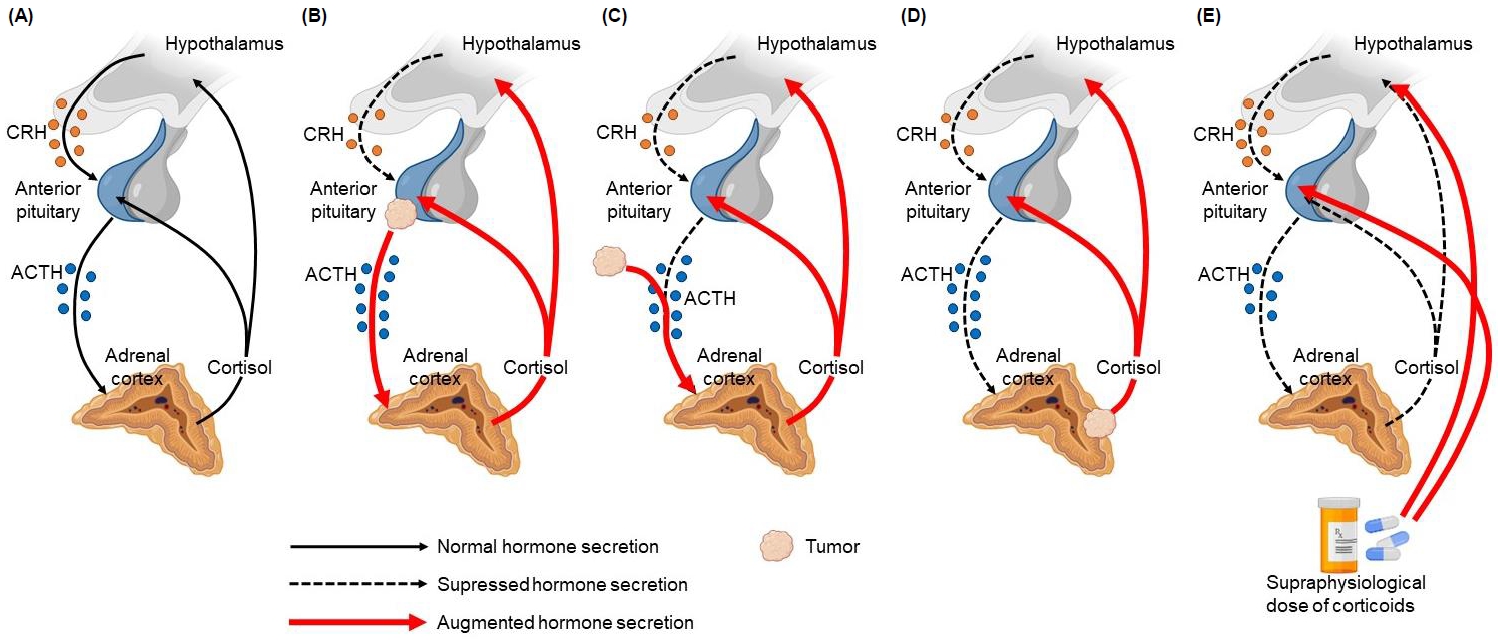
The hypothalamus produces corticotropin-releasing hormone (CRH), which is transported by the portal system to the anterior pituitary, where it stimulates the release of ACTH, which in turn regulates cortisol secretion by the adrenal cortex. Cortisol exerts negative feedback by inhibiting the secretion of CRH and ACTH. This is called the hypothalamic-pituitary-adrenal (HPA) axis [12]. In CS, normal regulation of the HPA axis is impaired because of the loss of negative feedback caused by excessive secretion of ACTH or cortisol (Fig. 1) [13]. Subtypes depend on the cause of the increase in cortisol secretion; thus, subtypes can be ACTH-dependent or ACTH-independent. In ACTH-dependent CS, the cause of increased cortisol secretion is hyperstimulation of the adrenal cortex by excess ACTH, which might originate from an ACTH-producing pituitary adenoma (CD) or from ectopic, nonpituitary ACTH secretion (ectopic ACTH syndrome), the latter usually secondary to a neuroendocrine tumor, such as small cell lung carcinoma or carcinoid lung tumors [1,12-14]. Regarding ACTH-independent CS, a benign adrenocortical adenoma, carcinoma, or rare forms of bilateral adrenal disease leads to excess cortisol secretion by the adrenal glands without ACTH stimulation. Exogenous CS represents the predominant etiology of CS in pediatric patients, which usually involves treatment with oral glucocorticoids in supraphysiological doses; however, it is also associated with intra-articular, intrathecal, epidural, inhaled, topical, or ocular administration route [1,10,12,13].
Several genetic mutations are responsible for syndromes associated with pituitary adenomas, including MEN1, which causes multiple endocrine neoplasia type 1; cyclin-dependent kinase inhibitor 1B; Guanine nucleotide-binding protein, α stimulating, which causes McCune-Albright syndrome; Aryl hydrocarbon receptor-interacting protein, which causes familial isolated pituitary adenoma; and ubiquitin-specific protease 8 (USP8), which is the most common mutation found in patients with CD and prevents lysosomal degradation of the epidermal growth factor receptor, increasing its deubiquitination, proopiomelanocortin transcription, and ACTH secretion [6]. In pediatric CD patients, the USP8 gene is mutated in 31%–63% of corticotroph adenomas, and these patients are more likely to experience recurrence after surgery [6]. However, the major driver mutations in USP8 wild-type tumors remain unknown.
There are other genetic mutations associated with CD. The RASD1 gene mutation can contribute to cell proliferation and ACTH secretion in a small subpopulation of cells [15]. TP53-inactivating mutations have been found in 3 CD cases [15]. N3CR1 mutations make the corticotroph cells unresponsive to negative feedback from the adrenal gland and resistant to the effects of glucocorticoids [15]. Activating mutations in the RET gene are associated with multiple endocrine neoplasia type 2, with pituitary adenomas having been reported in rare cases [15,16]. The "Three P Association" (3PAs) is the association of a pituitary adenoma with a pheochromocytoma or paraganglioma and could represent variants of multiple endocrine neoplasia or CD [15]. Loss-of-function CABLES1 missense mutations have been identified in 4 CD patients [15]. Loss-of-function mutations in the DICER1 gene are associated with various tumors, including pituitary blastoma, which presents clinically as CD early in infancy [15,16].
Clinical features
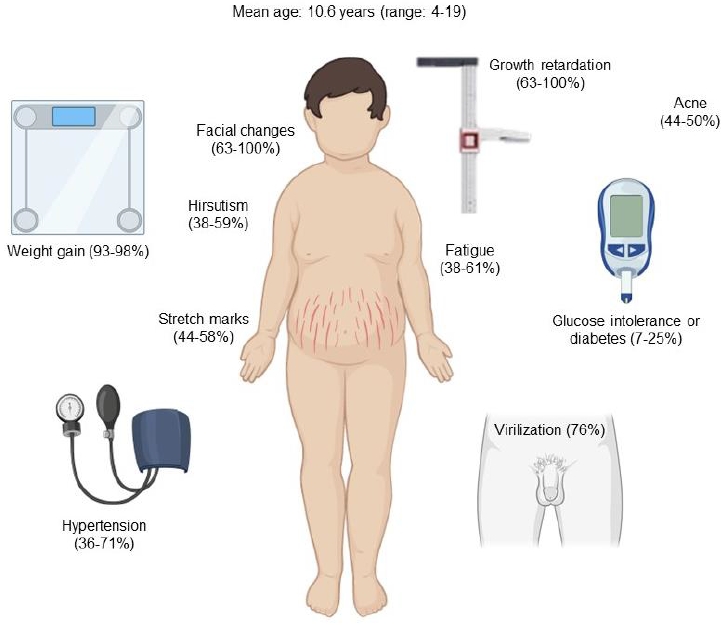
Early recognition of clinical signs and symptoms is essential for timely diagnosis and treatment [3]. The onset of the disease is usually insidious, delaying the diagnosis significantly. The mean duration of symptoms until diagnosis has been reported between 1.7 and 2.5 years [3,5]. Facial changes described as "Cushingoid facies" are very common in children and can be detected by comparison with previous photographs [3,17]. Growth retardation is another manifestation of chronic hypercortisolism and is usually associated with rapid or exaggerated weight gain and delayed bone age [5]. Increased adrenal androgens lead to abnormal virilization, such as acceleration of pubic hair and genital growth in boys with prepubertal testicular volumes or early pubic hair growth with prepubertal breast development in girls. Hirsutism, acne, and purple striae are also common in adolescents [3,5,17]. In children, the most suggestive features of CS are facial changes, rapid or exaggerated weight gain, hirsutism, virilization, and acne (Fig. 2). Mood disturbances, muscle weakness, osteopenia, and headache are less frequent symptoms [18].
Diagnosis
During diagnosis, ingestion or administration of exogenous corticosteroids should be excluded (oral, nasal, inhalation, topical, intramuscular, etc.) since exogenous CS is clearly more common than the endogenous form. At the initial assessment, the stage of sexual development based on the Tanner scale, bone age, and the presence of abnormal virilization should be determined [3].
- 24-hour urinary free cortisol (UFC): There is an intrapatient variability of approximately 50%; therefore, 3 consecutive samples (i.e., over 3 days) are recommended. In addition, a physiological increase in UFC excretion may occur in girls during the perimenarche phase, and false-negative results can occur in cases of kidney failure with creatinine clearance <60 mL/min [1,3,8]. The diagnostic cutoff is 24-hour UFC excretion > 70 μg/m2 (193 nmol/24 hr) [19].
- Midnight serum or salivary cortisol: Midnight serum cortisol should be collected with an indwelling IV, and a value greater than 4.4 μg/dL has high sensitivity and specificity for CS [1,17]. Late-night salivary cortisol has been shown to have superior diagnostic performance compared to UFC. However, there is great inter-laboratory variability [1]. Normal salivary cortisol level is <1.45 ng/mL (sensitivity 100%, specificity 95%) [23].
- Dexamethasone suppression test (DST): Characterized by failure of serum cortisol to decrease to <1.8 μg/dL [3]. This measurement is carried out by 2 techniques: (1) Overnight DST: Oral administration of 25 μg/kg (maximum dose 1 mg) dexamethasone at 23:00 PM or midnight, after which a serum cortisol sample is collected at 9:00 AM the next morning. (2) Low-dose DST: Oral administration of 20–30 μg/kg/day dexamethasone in children weighing <30 kg (maximum dose 2 mg/day) divided into 0.5 mg/dose every 6 hours (9:00 AM, 15:00 PM, 21:00 PM, and 3:00 AM) for 48 hours.
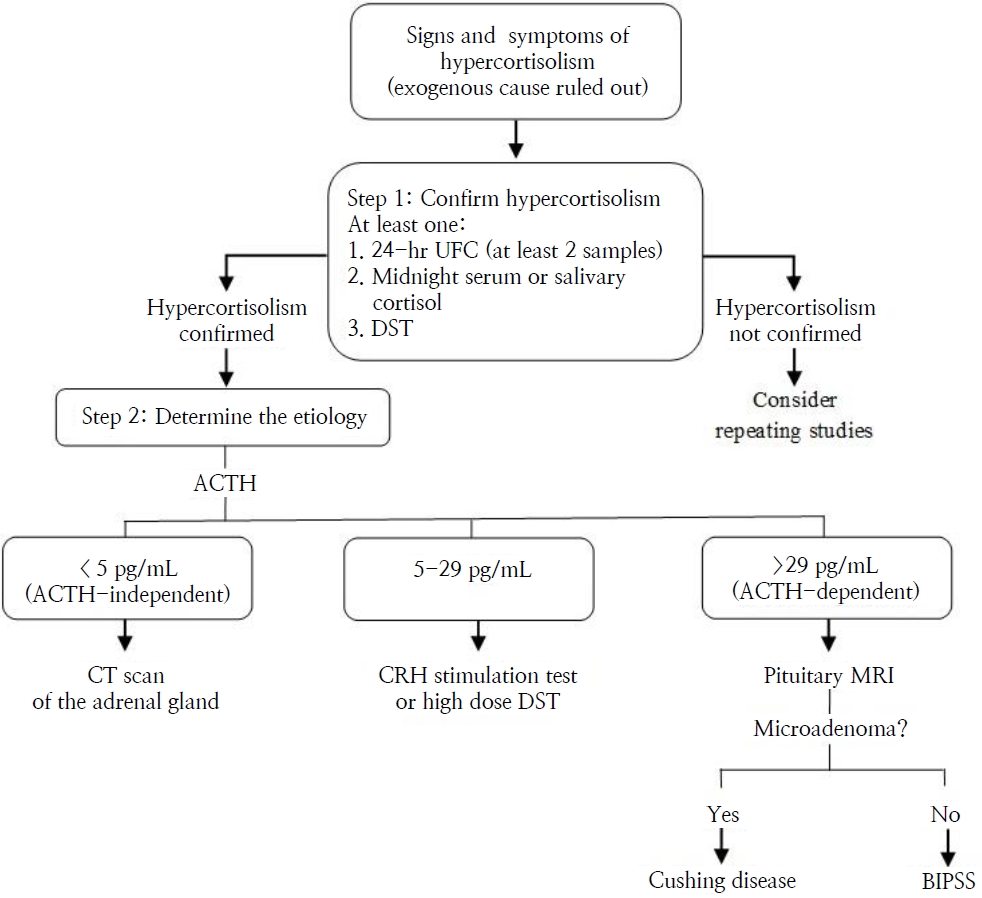
The next step is distinguishing ACTH-dependent from ACTH-independent syndrome (Table 2) [7,18,19,21,22,24] once the diagnosis of CS is confirmed:
- Basal ACTH: The ACTH cutoff points to establish whether CS is ACTH-dependent or -independent vary depending on the researcher. In this manuscript, our proposal is that a morning plasma ACTH >29 pg/mL is indicative of CD, whereas ACTH <5 pg/mL confirms primary adrenal disease [8,21].
- CRH or desmopressin stimulation test: Intravenous administration of 1-μg/kg CRH (maximum dose 100 μg) is recommended. An exaggerated (>20% from baseline) response of serum cortisol (at 30 and 45 minutes) and an at least 35% increase over basal values of ACTH (at 15 and 30 minutes) supported the diagnosis of pituitary-dependent hypercortisolism. Desmopressin testing can be used in patients with extremely difficult venous access or when CRH is not available [3].
- High-dose DST: The standard protocol either involves a single dose of 80–120 μg/kg (maximum dose 8 mg) dexamethasone administered orally at 23:00 PM or dexamethasone administration divided in 4 doses of 2 mg each for 48 hours. A decrease ≥20% in morning serum cortisol from baseline distinguishes CD from ectopic causes of ACTH production, with a sensitivity of 97.5% and specificity of 100% [1,3].
- Pituitary magnetic resonance imaging (MRI): Pediatric CD is mainly associated with corticotrophic microadenoma, usually <6 mm in diameter, generally hypodense on MRI, and frequently nonenhanced by gadolinium contrast. Therefore, it is necessary to perform thin-slice high-resolution MRI at CD specialist tertiary referral centers. Nevertheless, MRI detects CD pituitary adenomas in only 16%–71% of cases [8].
- Bilateral inferior petrosal sinus sampling (BIPSS) is generally reserved for pediatric patients who have confirmed ACTH-dependent CS and a negative pituitary MRI. Pituitary ACTH secretion is supported if there is a central-to-peripheral ACTH ratio >2 in basal conditions and >3 post-CRH stimulation, and samples should be obtained at 3, 6, and 10 minutes after administration of CRH or desmopressin (10 µg IV) [3,4]. The sensitivity of BIPSS in pediatric patients is lower than in adults but increases after desmopressin stimulation, proving that desmopressin is an efficient alternative to the CRH test.4) Lateralization of the adenoma to one side of the pituitary gland can be predicted if the interpetrosal sinus gradient is ≥1.4, guiding the surgeon during transsphenoidal surgery (TSS) [3,4].
Treatment
The goal of treatment is normalization of cortisol level and reversal of the signs and symptoms previously described [26]. Additionally, adequate growth and development must be preserved [27]. Different treatment options have been described for CD, and the choice depends on etiology, age, form of presentation, and availability of resources [28]. Management includes medication, surgical approaches, radiotherapy, or combination therapy [5]. Despite the increase of new treatments, achieving remission is still a challenge.
1. Surgical treatment
CD patients should receive surgical intervention as soon as possible. TSS with selective adenomectomy remains the first-line therapeutic intervention, wherein 2 techniques are available: microscopic and endoscopic, both allowing adequate visualization of the intrasellar content [29-31].
Microscopic TSS has been performed for a long time. TSS alone achieved a remission rate of 76%; in combination with adrenalectomy and/or radiotherapy, remission reached 91% in a series of 33 pediatric patients [31]. Early remission was achieved in 94.8% of patients in another series of 96 pediatric patients with CD who underwent microscopic TSS; however, 21.9% of cases recurred over time [32]. In addition, young age, high preoperative cortisol level, large tumor size, and cavernous sinus extension were positively correlated with recurrence [30].
Endoscopic TSS has been used for more than 10 years; however, there is a lack of experience described in the pediatric population. One of the first studies was performed in 27 children with pituitary adenomas, where total tumor resection was achieved in 81.5% of patients, and no postoperative complications, such as mortality, neurological morbidity, or late rhinological problems, were reported [34]. Clinical and biochemical recovery was achieved in 83% of patients in a study that included pediatric patients with CD, and there was no tumor recurrence after an average of 4.7 years [35].
Several studies have compared the 2 techniques. Remission of hypercortisolism was achieved in 88.23% of those who underwent endoscopic TSS compared to 56.6% of those who underwent microscopic TSS in a retrospective analysis of 104 patients [36]. In a recent systematic review and meta-analysis including 6,695 patients, 80% achieved remission of hypercortisolism, with no difference between techniques. However, the percentage of macroadenoma patients in remission was higher and the percentage of recurrence was lower after endoscopic TSS [37]. In the same study, cerebrospinal fluid leak was more frequent with endoscopic TSS, whereas transient diabetes insipidus was reported less often. However, these studies included pediatric and adult patients, and they did not differentiate the results based on age.
Bilateral adrenalectomy is becoming less common as a primary therapy. It is indicated when CD persists despite pituitary surgery or when rapid normalization of hypercortisolism is desired [38,39]. The reported complications are adrenal insufficiency (AI) and Nelson syndrome, which may develop in up to 25% of the pediatric population after follow-up based on studies from more than 30 years ago [40,41]. Currently, there are no efficacy data due to the very limited use of this procedure.
2. Radiotherapy
Pituitary radiotherapy (RT) was formerly used as a first-line therapy. Currently, it is indicated as a second-line therapy only in CD cases where surgery is not feasible [26]. It is recommended when there is recurrent CD and a second surgery is not possible [42]. It is also performed in patients with tumors exhibiting progressive growth and/or local invasion to the cavernous sinus, patients with Nelson syndrome, or prophylactically just after bilateral adrenalectomy to avoid tumor growth [38,43]. RT delivery techniques vary depending on whether the tumor is stereotactic or not and whether it is administered in small doses in several sessions or in a single dose. Radiation doses vary from 8 Gy to 45–50 Gy, delivered based on the size of the lesion, number of sessions, and type of RT [44]. Storr et al. [45] reported their experience with pituitary RT (45 Gy in 25 fractions) in 7 CD pediatric patients following unsuccessful TSS. RT was 100% effective, with no recurrences over time. Growth hormone deficit was reported as the main complication. Another similar study included 8 patients who underwent the same second-line pituitary RT regimen, and 4 patients (50%) were cured in a minimum follow-up of 2 years; 5 patients developed hypogonadism. In addition, all patients failed to reach their target height at the time of the last follow-up [46].
3. Medical treatment
If a tumor is identified, surgery is the first-line treatment, as discussed above. However, if surgery is not possible, pharmacological therapy is indicated.
Ketoconazole is an imidazole derivative primarily used in fungal infections [47]. Its action in steroidogenesis suppression (CYP17 and CYP11B1 enzyme inhibition) at adrenal and gonadal glands has been described for almost 4 decades [48,49]. The safety and efficacy of ketoconazole have not been established for pediatric patients younger than 12 years of age; thus, the main regulatory centers, such as the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA), have not proposed recommendations regarding its use and dosage in this population [50,51]. Case reports that described doses from 200 to 800 mg/day achieved serum and urinary cortisol normalization, as well as improvement of Cushingoid signs and growth velocity during follow-up [52,53]. Hepatotoxicity is the main safety concern and can occur in 0.5% to 4.2% of children and adolescents. Therefore, hepatic enzyme testing after 3 or 4 weeks of treatment is suggested [54]. Ketoconazole dosage reduction or withdrawal is required if hepatotoxicity occurs, and hepatic enzyme levels are expected to return to normal in an average of 3 weeks. 54)
Methyrapone is a pyridine derivative with an antisteroidogenic mechanism that results from enzymatic blockade of 11β-hydroxylase and 17α-hydroxylase, decreasing the production of glucocorticoids and mineralocorticoids [55]. In pediatrics, methyrapone is approved for diagnosis of AI and for treatment of endogenous hypercortisolism; however, the recommended therapeutic dose has not been established. Some independent studies have used doses from 720 mg to 1,500 mg/day [56-58]. The most frequent adverse effects are dizziness and mild gastrointestinal symptoms, usually within 2 weeks of treatment onset, and they typically resolve after drug discontinuation [59]. The safety and efficacy of this drug have been demonstrated in an 8-month-old infant with ectopic CS [56]; however, there are no reported cases in patients with CD.
Mitotane is a bimodal agent since it has an adrenolytic effect due to the cytotoxicity induced by mitochondrial damage. It also inhibits several enzymes, such as 11β-hydroxylase, 18-hydroxylase, and 3β-hydroxysteroid dehydrogenase, leading to a decrease in steroidogenesis [47]. Thus, it is not only restricted to hypercortisolism treatment, but is also used in adrenal carcinoma as a chemotherapeutic agent [60,61]. Growth velocity and pubertal development were restored in 9 pediatric patients with CD receiving mitotane at an initial dose of 1 g/day, regardless of hypercortisolism resolution [62]. The most frequent adverse effects are gastrointestinal issues. Additionally, since mitotane is also considered an endocrine disruptor, hypothyroidism and gynecomastia have been described [60].
Mifepristone is a glucocorticoid receptor blocker that reduces the peripheral effects of hypercortisolism; therefore, it does not decrease serum cortisol level [63]. Furthermore, serum cortisol level may increase and, if excessive, can induce a high mineralocorticoid response, which produces hypertension, hypokalemia, and peripheral edema [64]. Mifepristone efficacy is measured according to clinical features instead of laboratory improvement due to its mechanism of action. Although the recommendation for the general population is a maximum dose of 20 mg/kg/day, a standardized dose has not been established in pediatrics [63]. Its safety and efficacy were demonstrated in a 14-year-old adolescent with ectopic CS [65], but there are no reported cases in patients with CD.
Osilodrostat is a steroidogenesis inhibitor that was recently approved by the FDA in 2020 for adult patients in whom pituitary surgery is not possible and has been recommended by the EMA for treatment of endogenous CS of any cause [66,67]. There are no established recommendations for pediatric patients to date. Osilodrostat inhibits aldosterone synthase and adrenal 11β-hydroxylase enzymes, decreasing the production of both glucocorticoids and mineralocorticoids [68]. The main adverse effects are nausea, headache, and fatigue [69]. A multicenter clinical trial including 12 patients under 18 years old with CD is currently in phase 2. In the study, the pharmacokinetics, pharmacodynamics, and tolerability of osilodrostat are being evaluated [70].
The use of somatostatin analogs such as pasireotide or a dopamine agonist such as cabergolide has been described as a valid option in conjunction with TSS, RT, or in cases where there is a postsurgical remnant [1,71]. Some case reports of adolescents with CD (12-, 15-, and 17-year-old patients) achieved successful outcomes after the use of somatostatin analogs [72-74]; however, there is no strong evidence of the use of these drugs in pediatric patients.
Prognosis and follow-up
There is no consensus on CD remission criteria [75]. Postoperative morning serum cortisol is the most important marker in defining remission. Two or more measurements within 2 weeks after surgery (on the 5th and 14th postoperative day) are recommended [76,77]. A cortisol value <1.8 μg/dL is highly indicative of remission in the pediatric population [78,79]. There are no sufficient data regarding the predictive value of postsurgical plasma ACTH, UFC, or low-dose DST [75].
After successful surgery for CD, the rapid onset of AI usually indicates a good prognosis [80]. However, rapid reduction in cortisol exposure often results in glucocorticoid withdrawal syndrome (GWS) [81]. However, to date, there is no scientific literature on GWS syndrome in pediatric patients with CD. GWS occurs following withdrawal of supraphysiologic exposure to either endogenous or exogenous glucocorticoids over a duration of several months [82]. The clinical manifestations are indicative of cortisol deficiency, as evidenced by HPA axis suppression, even with supraphysiologic glucocorticoid replacement therapy. GWS symptoms typically occur 3–10 days postoperatively [81].
After CD surgery, once remission is achieved, exogenous glucocorticoid replacement should be initiated and maintained during the months required for HPA axis recovery [83]. The most important treatment is periodic assessment of the HPA axis until its recovery [84].
Recurrence of CS was variable in a case series, ranging from 7% to 25% at 5-, 10-, and 15-year postsurgical follow-up [85,86]. The following prognostic factors of pediatric CD recurrence have been described: older age at onset of symptoms, petrosal or dural sinus invasion, unsuccessful adenoma localization during surgery, larger tumor diameter, corticotroph adenomas with USP8 gene mutation, and corticosteroid replacement for less than 6 months after surgery [25,75,79].
Posttreatment challenges include optimizing pubertal growth and development, normalizing body composition, and promoting mental health and cognitive maturation [87].
Clinical recovery is slower and/or incomplete, and it is not rare to find persisting medical problems such as hypopituitarism, loss of final height, obesity, hypertension, osteoporosis/osteopenia, and cognitive impairment despite successful treatment of hypercortisolism (biochemical recovery) [88]. In a prospective study (up to 7-year follow-up) of 13 children and adolescents with CD who were successfully treated, a -0.8 standard deviation (SD) loss of final adult height and 0.9 SD increase in weight and body mass index were found despite no recurrence of CS [89]. Bone mineral density was more severely affected in the vertebra and was partially recovered after definitive treatment in another study of 35 children with CD [90].
Abdominal obesity and insulin resistance may persist after the treatment of CD [91]. Hypertension, diastolic blood pressure, and mean arterial pressure usually normalize within 3 months after surgery, whereas systolic blood pressure may take up to 1 year to normalize [92]. However, CD may persist with hypertension in up to 20% of patients after cortisol normalization [93].
CD has been associated with multiple psychological and psychiatric disturbances, with emotional lability, depression, and anxiety being the most common [1,3]. These disturbances may persist after remission of hypercortisolism and even after HPA axis recovery [94].
Table 3 lists the main long-term consequences of prolonged exposure to corticosteroids, as well as treatment-related complications.
Conclusions
CD is rare in the pediatric population and represents a challenge for physicians owing to its multiple associated conditions involving growth and pubertal development, body composition, cardiovascular status, bone mineral density, and psychological health; thus, it is important to achieve an early diagnosis, timely treatment, and to follow a multidisciplinary approach to control hypercortisolism and improve the prognosis and quality of life in pediatric patients.