Highlights
┬Ę Patients with partial androgen insensitivity syndrome (PAIS) exhibited various degrees of ambiguous external genitalia ranging from predominantly female to undervirilized male phenotypes. Molecular analysis of AR identified 19 mutations, including 7 novel sequence variants. Nonsense and frameshift mutations were frequent in patients with complete androgen insensitivity syndrome, whereas patients with PAIS harbored exclusively missense mutations.
Introduction
Androgen insensitivity syndrome (AIS) is an X-linked recessively inherited disorder caused by complete or partial unresponsiveness to androgens in individuals with a 46,XY karyotype because of mutations in the AR gene [1]. In 1953, Morris et al. first described testicular feminization syndrome in 82 females with bilateral testes, which was subsequently renamed "complete AIS (CAIS) [2]."
Patients with CAIS typically present with either inguinal hernia in the infantile period or primary amenorrhea in the adolescent period [3]. Development of estrogen-dependent secondary sexual characteristics, such as breast development, occurs as a result of excess aromatization of androgens; however, pubic or axillary hairs are usually absent or sparse. Partial AIS (PAIS) refers to phenotypes that include varying degrees of masculinization of the external genitalia because of partial androgen responsiveness, resulting in clitoromegaly, cryptorchidism, micropenis, hypospadias, or bifid scrotum [2]. For patients with PAIS, sex assignment is based on the external genitalia [4]. Due to an absence of the uterus, patients with AIS do not menstruate and are infertile. Mild AIS (MAIS) is generally associated with typical male genitalia in individuals presenting with gynecomastia or infertility in later life.5
The AR protein is a 110 kDa, 919ŌĆōamino acid protein composed of the following 4 major functional domains: the N-terminal domain (NTD) encoded by exon 1, the central DNA-binding domain (DBD) encoded by exons 2 and 3, a C-terminal ligand-binding domain (LBD) encoded by exons 4ŌĆō8, and a hinge region connecting the LBD to the DBD [2]. The NTD contains the activation function-1 region, which plays a pivotal role in transactivation of AR and regulates the interactions with the LBD [1]. The activation function-2 region is located in the LBD and is important for stabilizing the entire protein structure, allowing interaction between the NTD region and specific coregulators [6].
To date, >400 mutations in AR have been reported in the Androgen Receptor Gene Mutations Database (http://androgendb.mcgill.ca/). Among them, missense mutations are the most common, followed by nonsense mutations, deletions, insertions, and splice site mutations [7]. Nonsense mutations and small insertions and deletions causing frameshifts that lead to premature stop codons are more common in patients with CAIS than in those with PAIS [8]. Further, mutations that partially interfere with AR function manifest clinically as PAIS [1].
Materials and methods
1. Subjects
This study included 19 consecutive patients with AIS diagnosed from January 2004 to May 2022 at the Department of Pediatrics, Asan Medical Center, Seoul, Korea. Per the study inclusion criteria for AIS patients, eligible individuals had a 46,XY karyotype confirmed by molecular analysis of AR. AIS was classified as either CAIS or PAIS according to clinical manifestations. Patients who had complete female external genitalia with testes located in the inguinal or abdominal area or patients who presented with primary amenorrhea and breast development as well as sparse to absent pubic hair during puberty were classified as having CAIS. Patients with ambiguous genitalia, such as male-like external genitalia with cryptorchidism, micropenis, hypospadias, or female-like external genitalia with clitoromegaly, were classified as having PAIS [2]. Seven previously reported patients with AIS were included to allow delineation of the clinical and molecular spectra in the full cohort of AIS patients [12-14].
2. Clinical and endocrinological evaluations
Patient clinical parameters were retrospectively collected, including presenting features, sex of rearing, timing of gonadectomy, and pubertal outcomes. Height and weight were expressed as standard deviation scores based on Korean reference data [15]. Pubertal stage was rated according to the Marshall and Tanner criteria [16]. External genitalia were assessed using the Prader stage or external masculinization score [17].
Endocrine functions were analyzed, including serum luteinizing hormone (LH), follicle-stimulating hormone (FSH), estradiol, testosterone (T), and dihydrotestosterone (DHT). A human chorionic gonadotropin (hCG) stimulation test was performed in 12 patients to evaluate Leydig cell function. Serum T and DHT levels were measured at baseline and on the fourth day after consecutive intramuscular injections of hCG (IVF-C, LG Chemistry, Seoul, Korea) at 1,000 units/day for 3 days. Serum T level was measured by radioimmunoassay (Coat-A-Count, Diagnostic Products, Los Angeles, CA, USA). The response to hCG was regarded as positive if the T level increased to at least twice the baseline value [18]. Pelvic ultrasound or magnetic resonance imaging was performed to investigate M├╝llerian duct remnants.
3. Molecular analysis of the AR gene
The AR gene was sequenced by either Sanger (n=14), targeted gene panel (n=3), or whole-exome sequencing (WES) (n=2) methods. Genomic DNA was extracted from peripheral blood leukocytes using the Gentra Puregene DNA isolation kit (Qiagen, Hilden, Germany). For Sanger sequencing, 8 exons of AR and their exon-intron boundaries were amplified by polymerase chain reaction, using specific oligonucleotide primers. Polymerase chain reaction products were directly sequenced using an ABI3130x1 genetic analyzer (Applied Biosystems, Foster City, CA, USA), as previously described [12]. Target gene panel sequencing was performed using a customized panel including 67 genes previously reported as associated with disorders of sex development in humans, as previously described [13]. For WES, SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) was used for library preparation. Sequencing was performed using the NextSeq500 platform (Illumina Inc., San Diego, CA, USA), generating 2 ├Ś 150-bp paired-end reads. Sequence reads were aligned to the human reference genome (hg19) using the Burrows-Wheeler Alignment program (version 0.7.12, https://bio-bwa.sourceforge.net/). The SAMtools 0.1.19 and Genome Analysis toolkits (version 3.5, http://samtools.sourceforge.net/) were used for single-nucleotide polymorphism variant calling from aligned sequence reads. The Genome Analysis toolkit version 3.5, FreeBayes 0.9.2.1, and Scalpel-0.5.3 were used for insertionŌĆōdeletion variant calling. After removing duplicates with PICARD (version 1.96, https://broadinstitute.github.io/picard/), annotation was performed with Variant Effect Predictor19) and dbNSFP [20].
Sequence variants were classified according to the standards and guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [21], and those identified by targeted gene panel sequencing or WES were validated by Sanger sequencing.
Results
1. Clinical and endocrine characteristics of AIS at presentation and clinical outcomes
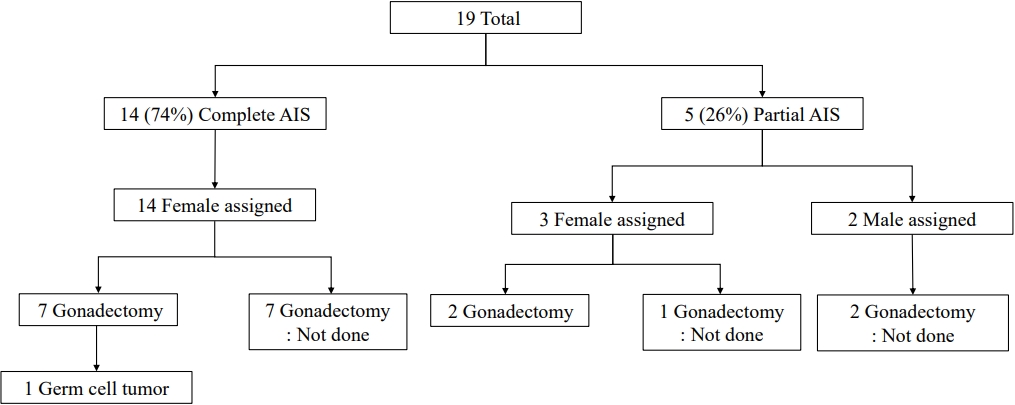
Among all 19 patients included in this study, 14 (74%) were classified as having CAIS, while the remaining 5 (26%) had PAIS. The clinical characteristics of the 19 patients with AIS are summarized in Table 1 and Fig. 1. The median follow-up duration was 4 years (range, 3 months to 17 years; Table 1). All patients with CAIS were raised as females. Among 5 patients with PAIS, 3 were raised as female, and the other 2 (patients 2 and 11) were assigned male at birth (Table 1). No patients have presented with gender identity problems to date.
Five patients with CAIS presented with primary amenorrhea at the age of 18.4┬▒4.2 years (range, 13ŌĆō25 years) and showed breast development to Tanner stage IIIŌĆōV; however, there was no pubic hair development. Among these 5 patients with CAIS, patient No. 4 manifested primary amenorrhea but did not visit the hospital. At the age of 30 years, she was referred to our institute because of abdominal pain and was diagnosed with a mixed germ cell tumor of the gonad, which required surgery and chemotherapy.
Seven patients with CAIS were brought to the hospital because of inguinal hernia at the age of 8.8┬▒6.7 months (range, 3 days to 19 months). Six of them (patient Nos. 5, 6, 10, 13, 16, and 18) underwent bilateral inguinal herniorrhaphy at the age of 9.1┬▒7.2 months (range, 1ŌĆō19 months), whereas the remaining patient (No. 12) was lost to follow-up.
The remaining 2 patients with CAIS presented during the neonatal period: patient No. 9 was referred because of palpable gonads in the labia majora, and patient No. 17, who had female external genitalia, was referred to our clinic because of a 46,XY karyotype detected by G-scanning (MGmed, Seoul, Korea), a genetic screening test performed to detect chromosomal abnormalities using a microarray. Both intra-abdominal testes were detected in patient No. 17 by magnetic resonance imaging.
Patients with PAIS presented at a median age of 2 years (range, 4 days to 3 years) because of clitoromegaly (n=3), ambiguous genitalia (n=1), hypospadias (n=2), micropenis (n=2), or inguinal hernia (n=1). Patient No. 1, who was raised as female, presented with clitoromegaly at 2 months of age and underwent gonadectomy and clitoroplasty at the age of 4 years. Patient No. 2 presented with hypospadias and inguinal gonads at the age of 4 days. Orchiopexy was performed at the age of 12 months, and repair of hypospadias was conducted at the age of 3.5 years. His stretched penile length was 2 cm at 5 years old. Two patients with PAIS (patient Nos. 7 and 15), who were reared as female, underwent bilateral inguinal herniorrhaphy during the infant period. Patient No. 11 presented with micropenis and hypospadias at the age of 3 years and underwent repair of hypospadias. He was lost to follow-up after surgery and visited the outpatient clinic at the age of 19 years because of delayed puberty and gynecomastia and has been treated with monthly testosterone enanthate since.
Following hCG stimulation, all 12 patients tested showed elevated serum T level compared to baseline (Table 2). Five patients with CAIS (patient Nos. 3, 4, 8, 12, and 16) showed high LH and FSH levels, suggesting hypergonadotropic hypogonadism; patient No. 12 was 3 months old when the hormone tests were conducted, while the remaining 4 patients (patient Nos. 3, 4, 8, and 16) were 19┬▒7.8 years old when the test was performed.
Nine patients (47%) underwent gonadectomy at a median age of 7 years (range, 5 months to 30 years), and 7 of them had CAIS. Four patients with CAIS (patient Nos. 3, 4, 8, and 19) underwent prophylactic gonadectomy after pubertal age (22.3┬▒6.3 years; range, 15ŌĆō30 years). Three patients with CAIS (patient Nos. 9, 13, and 17) underwent gonadectomy at a median age of 14 months (range, 5ŌĆō22 months). Patient No. 9 showed testes protruding toward the labia majora and underwent orchiectomy with labiaplasty at the age of 5 months. Patient No. 13 underwent gonadectomy during inguinal herniorrhaphy at the age of 14 months. Patient No. 17 underwent gonadectomy at the age of 22 months before being diagnosed with CAIS by WES. Two patients with PAIS (patient Nos. 1 and 7, both reared as female) underwent gonadectomy during clitoroplasty at 4 and 7 years of age, respectively.
Four patients with CAIS (patient Nos. 3, 8, 13, and 19) and 2 patients with PAIS (patient Nos. 1 and 7), who were reared as female, have been treated with estrogen replacement therapy since the median age of 14┬▒8.8 years (range, 11ŌĆō33 years).
Bone mineral density (BMD) analysis was performed in one patient (patient No. 3) at the age of 34 years, which revealed BMD z-scores of the femur neck and spine of -2.2 and -3.1 points, respectively. She has been treated with denosumab at a dose of 60 mg every 6 months.
2. Mutation spectrum of the AR gene and genotypephenotype correlations
Nineteen mutations were identified in the 19 patients, with no mutation hotspots (Table 3, Fig. 2). Missense mutations were the most common (12 of 19, 63%), followed by small deletions (n=3), nonsense mutations (n=2), an insertion (n=1), and a splice site mutation (n=1). Mutations were maternally inherited in 7 patients (37%), while the remaining 12 patients did not undergo family testing. Family testing was performed in 2 female siblings of patients 9 and 18 and revealed normal sequences of AR.
Twelve patients (63%) harbored previously known pathogenic or likely pathogenic variants, while novel sequence variants were identified in the remaining 7 patients (37%), including c.1337dup (p.L446Ffs*56), c.2440del (p.F814Sfs*9), c.2327T>A (p.M776K), c.2353T>G (p.C785G), c.1768+1G>A, c.2380G>T (p.E794*), and c.2182del (p.N728Tfs*61). Six of the novel variants were classified as pathogenic or likely pathogenic using American College of Medical Genetics and Genomics criteria (Table 3). The remaining novel variant (p.M776K) was a variant of uncertain significance; however, it was predicted to be deleterious by in silico prediction programs and not found in the Genome Aggregation Database (http://gnomad.broadinstitute.org/), suggesting that it was the pathogenic cause of AIS.
The patients with CAIS harbored 7 missense mutations, 3 small deletions, 2 nonsense mutations, 1 insertion, and 1 splice site mutation. Two mutations (p.Q76* and p.L446Ffs*56) were located in the NTD, 3 (p.R616del, p.R616H, and c.1768+1G>A) were located in the DBD, and 9 (p.N728Tfs*61, p.M776K, p.E794*, p.C785G, p.F814Sfs*9, p.F828L, p.R841C, p.R841H, and p.L882P) were located in the LBD. Meanwhile, all 5 patients with PAIS harbored exclusively missense mutations in the LBD (n=3) or DBD (n=2).
Discussion
Here, we describe the clinical outcomes and mutation spectrum in a relatively large cohort of patients with AIS in Korea. The major phenotypes observed among patients with CAIS in this study were inguinal hernia in infancy or primary amenorrhea during puberty. Patients with PAIS exhibited various degrees of ambiguous external genitalia ranging from predominantly female to undervirilized male phenotypes, which may be attributable to residual AR function.
As patients with AIS are resistant to androgens, basal T and LH levels tend to be elevated because of a lack of negative feedback inhibition from the anterior pituitary gland [2]. Further, FSH level is usually normal because testicular inhibin B secretion is not impeded [2]. In the present study, no significant differences in hormone levels, including those determined by hCG stimulation testing, were detected between patients with CAIS and PAIS, consistent with findings of a previous report [8].
Patients with CAIS are at an increased risk for gonadal cancer [22]. Decreased AR activity and abnormal gonadal location can influence the survival of atypical germ cells, leading to histopathologic changes [23]. Gonadal tumors are rare in pediatric populations, where malignancy rates are low, ranging from 0.8%ŌĆō2.0% [24,25]; however, rates up to 22% have been reported in adults with CAIS [26]. In patients with PAIS, the gonads can be preserved for endogenous hormone function or fertility potential; therefore, the incidence of germ cell tumor is significantly high (up to 15%) [27]. For patients with AIS raised female, bilateral gonadectomy is recommended to avoid virilization and reduce the risk of gonadal tumors [27,28]. In the consensus statement on the management of disorders of sex development, gonadectomy should be performed in patients with CAIS and PAIS; however, there is no recommendation for the timing of gonadectomy [25]. Recommendations have been made to perform gonadectomy after puberty in patients with CAIS to induce spontaneous puberty [2]. As the risk of gonadal germ cell tumor before puberty is very low, it is reasonable to delay gonadectomy until adulthood [22]. In the present study, seven of 14 patients with CAIS and 3 of 5 patients with PAIS did not undergo gonadectomy. However, these patients have remained under surveillance for gonadal germ cell tumors by annual ultrasound. After gonadectomy, sex hormone replacement therapy is required to induce secondary sexual characteristics and promote psychosocial well-being and bone health [3], and this therapy should be administered until the age of natural menopause, with regular monitoring of BMD. In patients who wish to preserve their gonads, annual ultrasonography or magnetic resonance imaging of gonads is recommended [8]. In the present study, only one patient with CAIS (patient No. 4) developed a mixed germ cell tumor at the age of 30 years as the first presenting feature. Hence, early diagnosis and regular monitoring of patients with AIS are critical to prevent malignant gonadal tumors.
Infertility is a major issue associated with AIS. AIS patients raised female are infertile because they do not have a uterus. Although fertility is challenging for individuals with PAIS reared as male, there are a few cases of successful fertility achieved in men with PAIS using assisted conception techniques [29].
In the present study, all 19 patients with AIS harbored private mutations. To our knowledge, there have been no reports on racial differences in the mutation spectrum. Missense mutations were the most common in this study, consistent with previous studies [1,7]. Splice site mutations and large deletions spanning several exons are typically observed in <10% of patients [1]. Most mutations causing both CAIS and PAIS are located in the LBD, followed by the NTD and DBD [7]. More than half of all AR mutations are located in the LBD, and the majority of these is missense mutations, whereas approximately 20% of mutations are located in the NTD [30]. Exon 1 is the largest exon of AR and has a GC-rich region, which makes it difficult to sequence. Therefore, the mutation frequencies in exon 1 could be underestimated, given that the length of the NTD totals more than half of the AR gene [30], suggesting that systematic sequencing of exon 1 is necessary. Mutations in the NTD are more frequent in CAIS, whereas defects in the LBD are more common in PAIS [8]. Almost all AR mutations in MAIS are found in the NTD; however, the number of AR mutations related to this phenotype is small [8]. Of mutations in the NTD encoded by exon 1, 71% are nonsense or frameshift mutations causing CAIS, while only 29% are missense mutations, which are primarily associated with PAIS or MAIS [1].
Two patients with CAIS (patient Nos. 6 and 12) and one patient with PAIS (patient No. 2) harbored a hemizygous mutation of AR inherited from their mothers, who were diagnosed with polycystic ovary syndrome (PCOS). The mutations were c.2482T>C (p.F828L), c.2353T>G (p.C785G), and c.1789G>A (p.A597T), respectively. Previous studies have suggested that heterozygous AR variants are associated with PCOS or hyperandrogenism [31]. Nucleotide changes in AR may promote PCOS progression [32]. In addition, the number of CAG repeats in the region of AR encoding the NTD is inversely correlated with AR activity, and patients with PCOS tend to have lower mean CAG repeat numbers than controls [33].
This study has several limitations. First, some clinical data and endocrinological findings, such as external masculinization score, family history, hCG stimulation test results, obesity, BMD, and quality of life, were not available for all patients because of the retrospective study design. In addition, the follow-up periods and intervals used to identify long-term outcomes differed among individuals. Second, molecular analysis of AR was performed using different methods, including Sanger sequencing, targeted panel sequencing, or WES, depending on availability. Nevertheless, we describe the clinical and molecular characteristics of a relatively large cohort of Korean patients with AIS.
In conclusion, this study describes data on the long-term clinical outcome and molecular characteristics of 19 Korean patients with AIS. Molecular analysis of AR identified 19 mutations, including 7 novel sequence variants. Nonsense and frameshift mutations were frequent in patients with CAIS, whereas patients with PAIS harbored exclusively missense mutations. Patients with PAIS manifested various degrees of masculinization of the external genitalia. Early diagnosis and regular follow-up, as well as timely sex hormone replacement therapy, are required to prevent gonadal cancer and to improve quality of life.