Clinical and molecular characteristics of infantile-onset diabetes mellitus in Egypt
Article information

Abstract
Purpose
In patients diagnosed with diabetes mellitus (DM) before the age of 12 months, there is an increasing recognition of diabetes caused by single-gene mutations, also known as monogenic diabetes of infancy or neonatal DM (NDM). This study aimed to classify patients at Alexandria University Children’s Hospital (AUCH) diagnosed with infantile-onset DM into type 1 DM (T1DM) or NDM and to detect differences in molecular characteristics of NDM patients at our center in comparison to other countries.
Methods
This retrospective/prospective observational study was conducted on 39 patients diagnosed with infantile-onset DM (age of onset ≤1 year) at AUCH from January 2003 to November 2020. The patients were divided into 2 groups according to age at the onset of DM: ≤6 months and >6–12 months. Molecular testing was done in patients diagnosed with DM at ≤6 months and those with negative autoantibodies.
Results
Twelve patients were diagnosed with DM at age ≤6 months and 27 patients were diagnosed between 6–12 months. Seventeen patients (43.6%) had T1DM, whereas 9 patients (23.1%) had genetically confirmed NDM, including 3 harboring novel mutations. The most common genetic causes of NDM were EIF2AK3 mutations (n=3), followed by KCNJ11 (n=2) and ABCC8 (n=2). Other mutations included SLC19A2 (n=1) and INS (n=1). Three patients with potassium ATP channel mutations were transferred from insulin to sulfonylurea treatment.
Conclusions
It is essential to identify patients with NDM clinically and confirm the diagnosis by molecular testing to distinguish them from T1DM as it helps in refining their management, predicting prognosis, and guiding genetic counseling.
Highlights
· This the first study in Alexandria, Egypt, addressing infantile-onset diabetes. We studied 39 patients diagnosed with diabetes mellitus (DM) at age of onset ≤1 year in Alexandria University Children’s hospital.
· Seventeen patients (43.6%) had type 1 DM (T1DM), whereas 9 patients (23.1%) had genetically confirmed neonatal DM (NDM). Three novel and 6 known mutations causing permanent NDM were identified, and most of them presented with DM ≤6 months of age.
· Wolcott-Rallison syndrome was the most common cause of NDM (33.3%) in our cohort. Three patients had KATP channel mutations, and were transferred from insulin therapy to oral SU with improved glycemic control, after several years of being misdiagnosed with T1DM.
Introduction
Type 1 diabetes mellitus (T1DM) is the most common diabetes that occurs in childhood [1]. The advent of molecular diagnostics has helped to confirm single-gene mutations in patients who were originally misdiagnosed as T1DM during infancy [2].
Monogenic DM is classified into neonatal diabetes mellitus (NDM) or monogenic diabetes of infancy and maturity-onset diabetes of the young (MODY). NDM usually presents before the age of 6 months, and rarely between 6 months to 1 year. Activating mutations in the genes encoding inward-rectifying potassium channel (Kir6.2) and sulfonylurea receptor (SUR1) subunits of the pancreatic beta (β)-cell (potassium inwardly rectifying channel subfamily J member 11 [KCNJ11], and ATPbinding cassette subfamily C member 8 [ABCC8]) are the most common genetic causes of NDM and can be treated with oral sulfonylurea (SU). Therefore, confirming a genetic diagnosis in these patients can lead to tremendous simplification and improvement in their clinical care [3].
To the best of our knowledge, there is a limited number of publications about NDM in Egypt and Arab countries. Hence, we sought to classify patients with infantile-onset DM at Alexandria University Children's Hospital (AUCH) according to their type of DM and to investigate differences in the molecular characteristics of NDM patients in our cohort compared to other countries.
Materials and methods
This was a retrospective/prospective observational study conducted on 39 patients with infantile-onset DM between January 2003 and November 2020 at AUCH, Alexandria, Egypt. The children studied were recruited from the diabetes clinic and the diagnosis of DM was made using the International Society of Pediatric and Adolescent Diabetes guidelines. Newlydiagnosed patients were enrolled with a follow-up period of at least 6 months. The participants were divided into 2 groups according to age at the onset of DM. Group 1 included patients diagnosed ≤6 months of age, and group 2 included patients diagnosed between the ages of 6–12 months. Patients were then subclassified into T1DM or NDM based on antibody levels and genetic testing. Patients with transient hyperglycemia due to parenteral glucose infusion, septicemia, stress, or drugs were excluded.
All patients enrolled in this study were subjected to detailed history taking including demographic data, age at DM onset, family and perinatal history, and consanguinity. A thorough clinical examination was done to detect any associated extrapancreatic features.
1. Laboratory investigations
Routine investigations were performed including complete blood count, liver, and renal function tests. Blood glucose level, venous blood gases, urine dipstick, and serum C-peptide using radioimmunoassay were done at onset. Glycated hemoglobin (HbA1c) was recorded at onset and the last clinical follow-up to assess glycemic control. Pancreatic autoantibodies including glutamic acid decarboxylase-65 (anti-GAD-65) antibodies (Abs) and anti-insulin Abs were evaluated by radioimmunoassay. Islet cell antibodies were estimated by indirect immunofluorescence using human pancreatic substrate.
2. Molecular analysis
Comprehensive genetic analysis was performed for patients presenting with DM ≤6 months of age and for patients diagnosed between 6–12 months with negative autoantibodies. Genomic DNA was isolated from peripheral blood samples using a Qiagen DNA Minikit. Genetic testing was performed by the Exeter Genomics Laboratory, Exeter, United Kingdom, as part of collaborative research work.
Initially, Sanger sequencing was carried out on genes that were the most common causes of permanent NDM (PNDM) including KCNJ11, ABCC8, and the insulin (INS) gene. In addition, eukaryotic translation initiation factor-2 alpha kinase-3 (EIF2AK3) testing was performed in patients born to consanguineous parents. This was followed by comprehensive testing using targeted next-generation sequencing (tNGS) of >30 known genes causing NDM in 9 patients for whom enough DNA was available. This was done by the analysis of the coding regions and conserved splice sites of the following genes: KCNJ11, ABCC8, INS, EIF2AK3, AGPAT2, BSCL2, CISD2, CNOT1, COQ2, COQ9, EIF2S3, FOXP3, GATA4, GATA6, GCK, GLIS3, HNF1B, IER3IP1, IL2RA, INSR, LPL, LRBA, MNX1, NEUROD1, NEUROG3, NKX2‐2, PDX1, PTF1A, RFX6, SLC2A2, SLC19A2, STAT3, WFS1, and ZFP57 (Supplementary Table 1). This assay can detect partial, whole gene deletions, and duplications as described previously [4]. In addition, the parents were tested for mutations to confirm the pattern of inheritance. Variants were classified using the ACMG/AMP guidelines as pathogenic, likely pathogenic, uncertain significance, likely benign, or benign [5]. Other genes causing transient NDM were not tested as all NDM patients in the present study had PNDM.
3. Sulfonylurea trial
A SU trial was conducted in patients suspected to have potassium ATP (KATP) channel mutations. Glibenclamide was started at a dose of 0.2 mg/kg/day and increased gradually by 0.2 mg/kg weekly until the discontinuation of insulin therapy while maintaining normal blood glucose levels.
4. Follow-up
All patients were followed during their hospital admission and afterward in the clinic, and their treatment and DM course were recorded.
5. Statistical analysis of the data
Data were analyzed using IBM SPSS Statistics ver. 20.0 (IBM Co., Armonk, NY, USA), and P-values of ≤0.05 were considered statistically significant.
Results
1. Demographic data and clinical history
The age of DM onset in the patients studied ranged from 0.5–12 months, with a mean of 7.9±3.8 months. Patients were divided into 2 groups according to their age at onset: group 1 diagnosed with DM ≤6 months of age (12 patients, 30.8%), and group 2 diagnosed between 6–12 months (27 patients, 69.2%). Patients were classified according to their type of diabetes (T1DM, n=17; NDM, n=9; or unidentified genetic mutation, n=13).
The median age of the patients at the last follow-up was 3.5 years, with a male to female ratio of almost 1:1. One-third of the patients diagnosed with DM ≤6 months were born to related parents; however, no significant difference in consanguinity rates was detected in comparison to the other group (33.3% in group 1 vs. 18.5% in group 2, P=0.416). Patients diagnosed at ≤6 months of age had birth weights significantly lower than the other group (median 2.7 vs. 3.2 kg, P=0.001). Four patients (33.3%) were born small for gestational age (SGA) with a birth weight more than -2 standard deviation (SD) below the mean for the corresponding gestational age (GA) and sex (Table 1).

Demographic data, history, and initial presentation of the studied patients
2. Clinical presentation
The majority of the patients (92.3%) were initially diagnosed with diabetic ketoacidosis (DKA), while 2 patients presented with polyuria, and poor weight gain, and 1 patient was asymptomatic and diagnosed incidentally. Associated extrapancreatic features are illustrated in Fig. 1. Half of the patients in group 1 were short (height >-2 SD below the mean for age and sex), with a statistically significant difference between the 2 groups (P<0.001). Ten patients (25.6%) had associated neurological manifestations (4 patients, 33.3%, diagnosed ≤6 months vs. 6 patients, 22.2%, diagnosed 6–12 months, P=0.693). The most common neurological feature was epilepsy detected in 8 patients (20.5%). Three patients aged below 6 months at DM onset (25%) were diagnosed with Wolcott-Rallison syndrome (WRS) and had recurrent attacks of ahepatitis, which was precipitated by stress or infection (elevated liver enzymes, with or without jaundice, hepatomegaly, and abnormal coagulation profile), with a statistically significant difference in comparison to the other group (P=0.024).
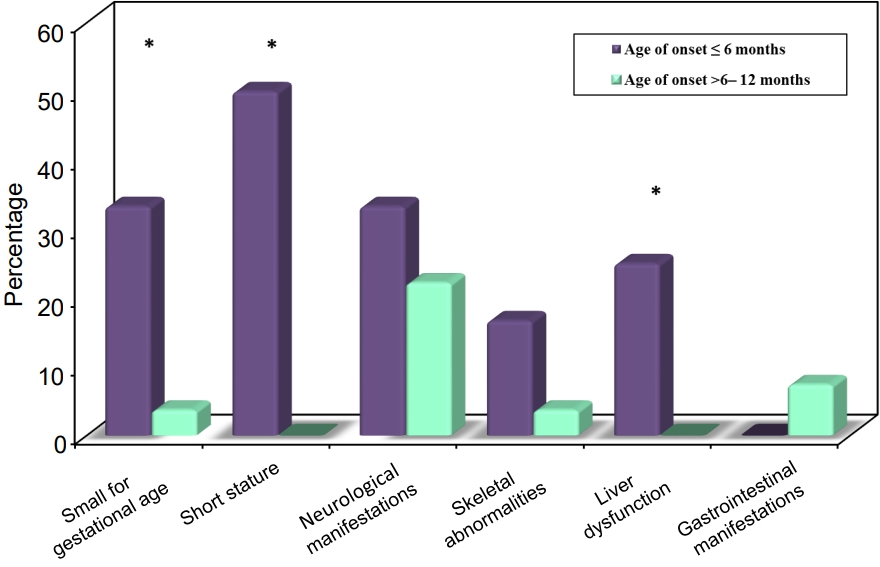
Comparison between the 2 ages of the onset groups according to associated extrapancreatic features.
3. Laboratory investigations
Laboratory investigations including random blood glucose, pH, C-peptide, HbA1c, anti-GAD-65, and anti-insulin antibodies are shown in Table 2. All patients presenting with DKA had positive urinary ketones (≥2+) by urine dipstick. Antiislet cell antibody was negative in all patients. Anti-GAD-65 antibody was positive in 51.9% of the patients diagnosed >6 months, whereas only one patient in group 1 (8.3%) was positive (P=0.013).
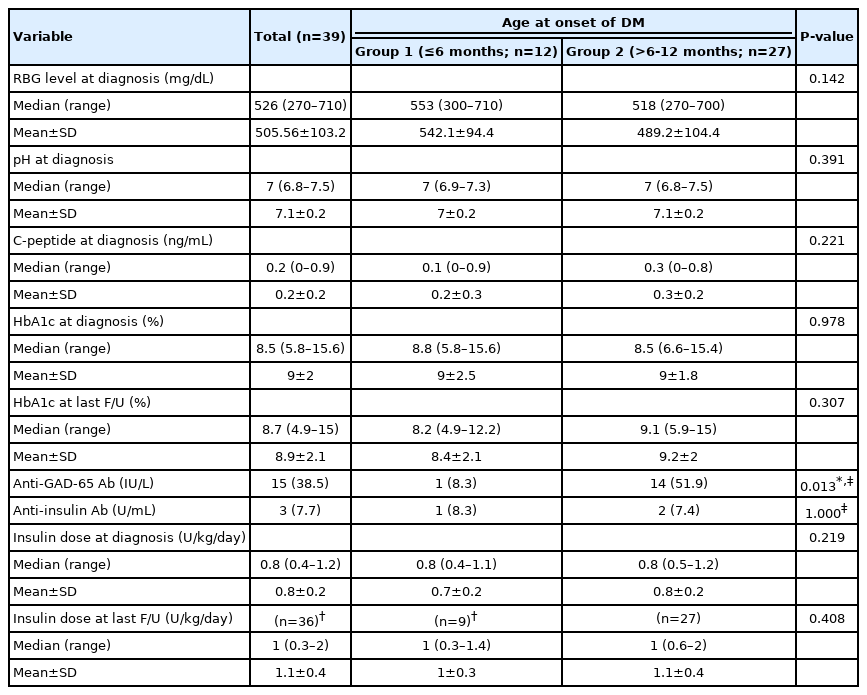
Comparison between the 2 studied groups according to laboratory investigations, and treatment
4. Genetic testing
Causative genetic mutations were detected in 9 out of 39 patients (23.1%), including 3 novel and 6 known mutations. A genetic diagnosis was confirmed in 7 patients (58.3%) in group 1 and 2 patients (7.4%) in group 2 (P=0.001) as illustrated in Fig. 2. A genetic mutation could not be identified in 13 infantile-onset diabetic patients; 3 of them were likely to have autoantibodynegative T1DM (T1bDM) with family members diagnosed with T1DM together with autoimmune thyroid diseases. T1DM was diagnosed in 17 patients (43.6%) with positive pancreatic autoantibodies; most of them were in group 2 (55.6%, 15 of 27 patients). The type 1 DM genetic risk score (T1D-GRS) was calculated in only one patient diagnosed with diabetes at 11 months with negative pancreatic autoantibodies and found to be 11.3 (75th percentile) confirming early-onset T1DM [6].
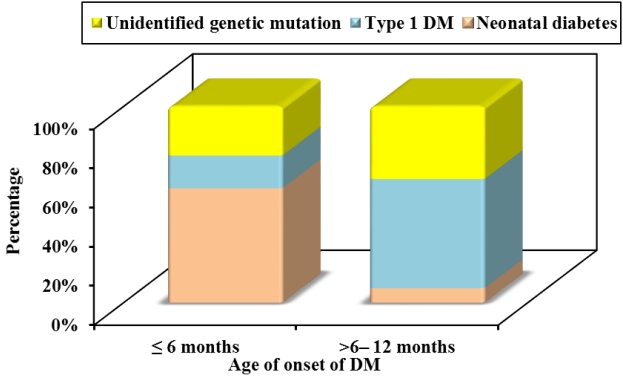
Cause of diabetes mellitus (DM) in the studied patients according to the age of onset.
The clinical and molecular characteristics of the patients with a confirmed genetic diagnosis are summarized in Tables 3 and 4. The most common genetic cause of DM in our cohort was EIF2AK3 mutations causing WRS detected in 3 patients (33.3%), all born to consanguineous parents. Two of the identified EIF2AK3 mutations in the present study were known, while a homozygous novel missense mutation [c.3191G>A, p.(Arg1064Gln)] was identified in 1 patient. This variant is not listed in the GnomAD database, it affects a highly conserved residue located in the protein kinase domain of the EIF2AK3 protein and was classified as likely to be pathogenic.

Clinical characteristics of patients with neonatal diabetes mellitus (n=11)

Molecular characteristics of patients with neonatal diabetes mellitus (n=11)
Activating mutations in the potassium ATP (KATP) channel genes were the second most common cause of NDM with 2 patients having mutations in KCNJ11 (22.2%) and 2 in ABCC8 (22.2%) as demonstrated in Fig. 3. Three patients with KATP channel mutations started a trial of SU while awaiting the results of their genetic testing and were successfully transferred from insulin to oral SU within 2 months. SU was started at a dose of 0.2 mg/kg/day and increased gradually according to blood glucose levels. The doses of SU ranged from 0.5 to 1.29 mg/kg/day. The 2 probands with KCNJ11 gene mutations required a lower dose of SU and a shorter time to switch to SU than the one with ABCC8 gene mutation as shown in Table 5. A patient diagnosed with DM at 7 months was found to have a novel heterozygous ABCC8 genetic variant of uncertain significance [p.(Gly1256Ser)] and was planned to start a trial of oral SU. Unfortunately, the parents refused and the patient was noncompliant.
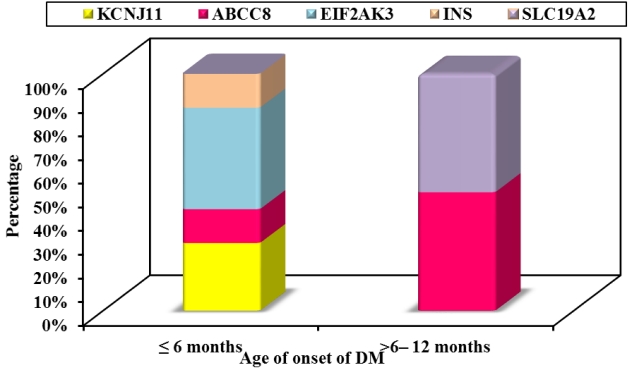
Neonatal diabetes mellitus (DM) subtypes in the studied patients according to the age of onset.

Neonatal diabetes mellitus patients with a successful switch to oral SU
Other genetic mutations identified in the current study included a de novo INS pathogenic variant in a patient diagnosed with DM at 2 months and a homozygous SLC19A2 mutation causing thiamine-responsive megaloblastic anemia (TRMA) in one patient who had pancytopenia with elevated reticulocyte count (5%) and negative Coombs test. She was diagnosed with stroke and DM at the age of 12 months (random blood sugar 300 mg/dL and elevated HbA1c 7% even though she had hemolytic anemia). In addition, she had bilateral optic atrophy and sensorineural hearing loss. Bone marrow biopsy stained with Prussian blue stain showed a picture of sideroblastic anemia. After thiamine therapy, her hematological findings, and HbA1c (5.8%) normalized, and her insulin requirements decreased from 1 to 0.7 U/kg/day.
Two patients were diagnosed with extremely early-onset T1DM at the age of 6 months. One patient with positive anti-GAD-65 Ab titer of 2,000 IU/ml (normal <10) had 5 siblings diagnosed with T1DM, whereas the other patient was positive only for anti-insulin Ab and had celiac disease. Three patients (25%) in group 1 had negative pancreatic autoantibodies, yet no genetic mutation causing their DM could be identified. One of these patients is likely T1bDM and has a history of maternal autoimmune thyroid disease and his paternal uncle has T1DM.
Discussion
The increased availability of genetic testing has allowed increased focus on the molecular basis of infantile-onset DM, with a single-gene mutation causing DM in some patients rather than autoimmunity. Monogenic DM patients (NDM and MODY) are often misdiagnosed as having T1DM, however, their pathogenesis, management, and prognosis differ.
Twelve patients (19.4%) in our cohort were diagnosed with DM within the first 6 months of life and were most likely to have a genetic mutation. NDM diagnosed before 6 months of age is rare (1:100.000 live births) in comparison to the incidence of T1DM, which is commonly diagnosed after the age of 6 months. Indeed, Rubio-Cabezas and Ellard [7] reported that the age at DM onset can be considered a cornerstone in diagnosing NDM, as most infants diagnosed before 6 months are less likely to be T1DM [8].
In the present study, we have included all patients diagnosed before 1 year so as not to miss the rare monogenic diabetes cases diagnosed >6 months (e.g., INS gene mutations) [9,10]. The median age at DM onset in our cohort was 9 months, which is higher than in other studies, such as Abujbara et al. [11] on PNDM patients in Jordan where the median age was younger (4–8 weeks) because only patients diagnosed <6 months were included.
Rare genetic conditions with autosomal recessive inheritance are common in consanguineous pedigrees [7]. This can potentially explain the higher percentage of patients born to consanguineous parents in group 1 (33.3% vs. 18.5% in group 2) as they are more likely to have a monogenic cause of DM, which can be inherited in an autosomal recessive manner such as WRS. The rate of consanguinity in our cohort was lower than in similar studies. For example in a study by Abujbara et al. [11], consanguinity was present in 45.5% (n = 10) of the patients, whereas other studies in Arab countries and a Turkish cohort showed that the majority of patients (89%) were born to firstdegree consanguineous parents [12,13].
Iafusco et al. [14] studied the clinical, genetic, and epidemiological features of patients diagnosed with diabetes in the first year of life in Italy and reported a higher percentage of preterm neonates (GA<37 weeks) among patients diagnosed before 6 months of age compared to those diagnosed after (15.4% vs. 2.7%, P=0.06). We did not see a significant difference in our cohort with a median GA of 39 weeks in both age groups.
Patients diagnosed ≤6 months of age had birth weights significantly lower than the other group (P=0.001). This is consistent with the majority of individuals in this group having a genetic etiology (such as mutations in KCNJ11, ABCC8, INS, and EIF2AK3), likely resulting in insulin deficiency in utero and affecting intrauterine anabolism and growth. This is similar to reports from other NDM cohort studies [11,14].
The probability of having DM with a monogenic cause was higher in patients who were born SGA, as 4 out of 5 patients with SGA were diagnosed with NDM ≤6 months of age and had a confirmed genetic diagnosis (33.3% of group 1). Similarly, Iafusco et al. [14] reported that two-thirds of infants diagnosed with DM in the first 6 months were born SGA, in contrast to 15% of those presenting at 6–12 months (P=0.001). Moreover, Johnson et al. [15] reported that infants diagnosed with DM at <6 months of age with high T1D-GRS had low birth weights compared with the World Health Orgnization international reference population (median z-score, -0.89 SD, n=48).
All patients diagnosed with DM ≤6 months presented with DKA, which might have been due to a delay in diagnosis due to the difficulty of interpreting the symptoms in this age group. This is in agreement with Letourneau et al. [16] who studied 88 cases with DM presenting <13 months of age in the University of Chicago Monogenic Diabetes Registry and reported a high risk of DKA with an overall frequency of 66.2%. We did not detect a statistically significant difference in rates of DKA presentation in the current study, which could be explained by the smaller number of patients (n=39).
The median HbA1c at diagnosis was 8.8% in patients diagnosed ≤6 months of age, even though hemoglobin (Hb) F is more prevalent than HbA at this age. Similarly, the median HbA1c in patients with NDM in Turkey was 10.2% (5.8%– 17.1%) in the study by Öngen et al. [17]
In the present study, 15 patients (38.5%) were positive for anti-GAD-65 Ab and 3 patients were positive for anti-insulin Ab (7.7%). Most of the patients with positive anti-GAD-65 Ab were diagnosed between 6–12 months (14 patients, 51.9%), with a statistically significant difference in comparison to those diagnosed ≤6 months of age (P=0.013). This is similar to the study by Huopio et al. [18] reporting positive DM-associated autoantibodies in 40% of patients diagnosed with early-onset DM during the first 6 months of life versus 70.8% of those diagnosed between 7–12 months in Finland.
We identified 3 novel and 6 known mutations causing PNDM in our cohort. More than 75% of patients with a genetic diagnosis presented with DM ≤6 months of age. WRS caused by EIF2AK3 gene mutations was the most common cause of NDM accounting for 42.9% of patients diagnosed at age ≤6 months. This was similar to that reported in the study by Asl et al. [19] with 28 of 124 Iranian NDM cases (22.58%) having a recessive mutation in EIF2AK3. On the other hand, Öngen et al. [17] reported that ABCC8 was the most common mutation causing NDM in their Turkish cohort (6 of 16 patients), followed by KCNJ11 and EIF2AK3 gene mutations (3 of 16).
The 3 patients with WRS in our cohort were born to consanguineous parents, who were confirmed to be heterozygous carriers for the EIF2AK3 pathogenic variant. Two of those patients had siblings diagnosed with NDM who died of fulminant liver failure during infancy. WRS patients in the present study had variable phenotypic characteristics including hepatomegaly, neutropenia, nonautoimmune hypothyroidism, and epiphyseal dysplasia. They all had short stature and recurrent attacks of liver dysfunction. In consanguineous families with a high level of homozygosity, PNDM is commonly associated with syndromic forms of DM such as WRS which has been reported to be the most frequent cause. [7,20].
The second most common cause of NDM in our cohort were KATP channel mutations, which can respond to oral SU that acts on SUR1 in the pancreatic β cell, leading to the closure of the KATP channel and causing membrane depolarization. This eventually causes calcium influx, and insulin secretion explaining the increase in C-peptide levels after SU treatment. [21]
Three of the 4 patients with KATP mutations (2 patients with KCNJ11 and 1 with an ABCC8 mutation) were transferred from insulin therapy to oral SU (glibenclamide) at an age of more than 8 years, with improved glycemic control and normalization of fasting C-peptide levels. One patient was referred to our hospital at the age of 8 years, and 2 patients were followed up in our diabetes clinic. One of the patients had a failed trial of transferring to SU during infancy, whereas the other was the eldest patient with NDM in the current study and was diagnosed 17 years ago when molecular testing for NDM was not as advanced. None of the patients reported side effects from glibenclamide tablets or attacks of hypoglycemia. This is in concordance with a study by Bowman et al. [22], a large cohort of PNDM patients with KCNJ11 mutations were given a high SU dose, resulting in better glycemic control for at least 10 years.
KATP channels are also expressed in the brain. Therefore, treatment with SU can also improve neurological manifestations in patients with developmental delay, epilepsy, and neonatal diabetes syndrome caused by the KATP channel mutations. [23] None of the KATP channel mutations identified in this study are commonly associated with a neurological phenotype. However, one patient with a KCNJ11 mutation had cerebral palsy (hemiplegic type) due to hypoxic-ischemic insult at birth. Another patient diagnosed with an ABCC8 mutation had epilepsy, global developmental delay, muscle weakness, and squint, and his brain imaging showed left occipitoparietal vascular malformation with cortical and subcortical hemorrhagic lesions, and gyral calcification of the left parietal lobe even though the ABCC8 gene mutation is not commonly associated with neurological symptoms. It was difficult to assess if neurological improvement occurred with SU as the treatment started years after diagnosis.
A heterozygous dominant INS gene mutation was identified in one patient, who had no extrapancreatic manifestations as INS is mostly expressed in pancreatic β-cells. [10]. Mutations in the INS gene lead to misfolding of the proinsulin protein resulting in increased endoplasmic reticulum stress, slow progressive β-cell destruction, and eventually apoptosis [24].
One patient was diagnosed with TRMA and had a family history of optic atrophy and DM. This highlights the importance of history taking in the interpretation of NDM subtypes and how crucial it is to reach an accurate diagnosis for the genetic counseling of the family.
This is the first study in Alexandria, Egypt, addressing infantile-onset DM and identifying patients with monogenic DM, including novel and known NDM gene mutations. Three of our patients stopped insulin and were transferred to oral SU after several years of being misdiagnosed with T1DM. However, our study has some limitations. The present study included a small number of patients diagnosed ≤6 months of age (n=12). Seven patients were tested for pancreatic autoantibodies after 5 years of diagnosis with DM. Hence, negative values might be due to the waning of antibodies or T1bDM rather than a monogenic cause. Other autoantibodies (islet antigen 2 antibody and zinc transporter 8 antibody) were not tested as they were not available. In addition, it was not possible to conduct a tNGS analysis of all patients with negative antibodies due to the limited availability of DNA.
In conclusion, the identification of patients with NDM either clinically or by molecular testing and distinguishing them from T1DM is essential as it helps in refining their management, predicting their prognosis, and determining the risk of recurrence. Our study highlights the importance of genetic testing, even years after the initial diagnosis. Similar to other Arab countries, WRS is the most common genetic mutation identified in our center in Alexandria, Egypt, despite the prevalence of consanguinity being lower in our cohort.
Ethical statement
of Alexandria University Ethical Committee on 31/12/2018 (number: 0201183), and informed consent from parents or legal guardians of all patients included in the study was obtained.
Supplementary Materials
Supplementary Table 1 can be found via https://doi.org/10.6065/apem.2142184.092.
Supplementary Table 1.
Gene symbols and nomenclature
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Data availability
The data that support the findings of this study can be provided by the corresponding author upon reasonable request.
Author contribution
Conceptualization: YA, EM, SE; Data curation: YA, EM, SE; Formal analysis: YA, EM, SE; Methodology: YA, IM; Project administration: EM; Visualization: EM, IM; Writing - original draft: YA; Writing - review & editing: YA, IM, EDF, SE
Acknowledgements
We are grateful to Prof. Dr. Sian Ellard, Prof Andrew Hattersley, and Exeter University, Exeter, UK, for their work and collaboration in the genetic analysis of our patients and we thank Prof. Dr. Magdy Omar, Professor of Pediatrics, Alexandria Faculty of Medicine, for his continuous support.