Adrenocortical carcinoma and a sporadic MEN1 mutation in a 3-year-old girl: a case report
Article information

Abstract
Childhood adrenocortical carcinoma (ACC) is a rare disease that is mostly linked to familial cancer syndrome. Although the prevalence of ACC is extremely low in children, it is clinically important to diagnose ACC early because age and tumor stage are closely related to prognosis. From this perspective, understanding the underlying genetics and possible symptoms of ACC is crucial in managing ACC with familial cancer syndromes. In this report, we present the case of a 3-year-old girl who initially presented with symptoms of precocious puberty and was later found to have ACC by imaging analysis. On genetic analysis, the patient was found to have a MEN1 gene mutation. MEN1 mutations are found in patients with multiple endocrine neoplasia type 1 (MEN1), usually precipitating multiple endocrine tumors, including pituitary adenoma, parathyroid hyperplasia, and adrenal tumors. Although MEN1 mutation is usually inherited in an autosomal dominant manner, neither of the patient’s parents had the same mutation, making hers a case of sporadic MEN1 mutation with initial presentation of ACC. The clinical course and further investigations of this patient are discussed in detail in this report.
Highlights
· Multiple endocrine neoplasia type 1 is a rare syndrome involving multiple endocrine organs. Clinical importance of this hereditary syndrome is addressed through this case report, a first report of childhood adrenocortical carcinoma with MEN1 mutation in Korea.
Introduction
Multiple endocrine neoplasia type 1 (MEN1), first identified in the 1900s, is a rare, hereditary syndrome characterized by multiple endocrine and non-endocrine tumors [1]. Genetically, MEN1 is inherited in an autosomal dominant manner, showing a high degree of penetrance. By inactivating mutations in the tumor suppressor gene MEN1, this syndrome manifests multiple endocrine problems such as parathyroid hyperplasia, pituitary adenoma, and tumors that originate from the pancreas and adrenal glands. By age 40 years, almost 90% of patients with MEN1 develop parathyroid hyperplasia and/or tumors, 40% have pituitary adenomas, and 70%–80% develop tumors that originate from the pancreas or gastrointestinal organs [2]. Data of young patients with MEN1 are limited since this syndrome is rare and manifests mainly in adults. A cohort study of 924 young patients with MEN1 reported that 17% developed MEN1-associated tumors before 21 years of age, mostly diagnosed after 10 years of age with increasing disease penetrance [3]. Among them, only 2 patients had adrenal tumors, with very low disease penetrance in the adrenal gland in children.
With regard to adrenal tumors, including adrenocortical carcinoma (ACC), approximately 80%–90% of children with adrenal tumors have hyperfunctioning adrenal glands, producing excess sex and steroid hormones such as cortisol, aldosterone, and testosterone [4]. Patients with ACC commonly have cushingoid features such as central obesity and a moon face. Due to excess testosterone, female patients with ACC may experience virilization with ambiguous genitalia and hirsutism. Patients seldom have hypertension with increased aldosterone level. Sometimes, tumors are detected due to increased size, causing abdominal pain by occupying the peritoneal space. Adrenal tumors are usually detected by ultrasonography, computed tomography (CT), or magnetic resonance imaging (MRI), although the final diagnosis of ACC can only be made on biopsy. ACC tends to occur in several familial cancer syndromes, and specific gene mutations in TP53, MEN1, PRKAR1A, and APC are well-known causes of ACC [5]. The TP53 (on chromosome 17p13.1) mutation is responsible for 50%–80% of ACC in children [6]. In southern Brazil, children with this mutation have a 15-fold increased incidence of childhood adrenocortical tumors [7].
A relatively low incidence rate has been reported in adrenocortical tumors (<40%) compared to other endocrine organ tumors in MEN1 [2]. Among adrenal tumors in MEN1 patients, ACC is rarer in children; however, early detection of ACC is important because it is characterized by aggressive malignancy. Approximately 10% of MEN1 patients have adrenocortical tumors, and only 14% of these adrenocortical tumors are malignant.
To date, there have been 15 cases of MEN1 in Korea, among which only one study reported MEN1 in a child [8]. The current case report describes a 3-year-old girl who presented with precocious puberty due to functional ACC. Upon genetic investigation, although the patient was found to have a MEN1 gene defect, neither of her parents had the same mutation. In this case report, the clinical course of the 3-year-old patient with de novo MEN1 mutation is presented, and the significance of the findings are discussed.
Case report
A 3-year-old girl visited the endocrinologic outpatient clinic in our pediatric department with a chief complaint of precocious puberty. She had acne, early developmental pubic hair, and her voice had started to deepen 3 months before the initial visit. She was born at a gestational age of 40 weeks, weighing 3.5 kg, and had no perinatal problems. The patient experienced no other medical issues before her initial visit to the hospital. The patient's parents also had no medical issues. Heights of her father and mother were 177 cm and 168 cm, respectively.
Anthropometric and physical examinations were performed during the first visit. The patient's blood pressure and heart rate were within the normal reference range. The patient's height was 109.3 cm (>97th percentile, +3.12 standard deviation [SD]) and body weight was 21.2 kg (>97th percentile, +3.95 SD) [9]. The acneiform eruption was in the forehead area. Although the patient did not present with breast budding, there were several strands of pubic hair observed. The patient had suspiciously ambiguous genitalia with clitoral enlargement (Prader grade 2). The vagina was observed, and there was no palpable testis in the genitalia. No palpable mass was observed in the abdomen.
Hormonal and chromosomal studies were conducted to evaluate precocious puberty and ambiguous genitalia. Although biochemical assessment revealed normal electrolytes, the patient had pronounced elevation of adrenal androgens (Table 1). Serum testosterone was 3.22 ng/mL (reference range [RR], 0.02–0.1 ng/mL) and serum dehydroepiandrosteronesulfate (DHEA-S) was 1,469 μg/dL (RR, <65 μg/dL). Elevated levels of serum 17-hydroxyprogesterone (17-OHP) (4.11 ng/mL; RR, 0.03–0.9 ng/mL) and serum androstenedione (>10.10 ng/mL; RR, <0.42 ng/mL) were also observed. Conversely, follicle-stimulating hormone, luteinizing hormone, adrenocorticotrophic hormone, and cortisol levels were within the normal range with respect to sex and age. Chromosome analysis revealed a normal female karyotype, i.e., 46,XX.
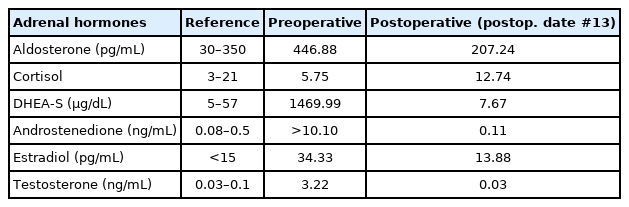
Changes in adrenal hormones level before and after surgery of adrenocortical carcinoma
Radiologic findings showed advanced precocious puberty. Her bone age was 11 years, preceding her chronological age by nearly 8 years. Abdominal sonography revealed a masslike lesion in the left adrenal gland (Fig. 1A), and MRI showed a 6.1×5.3×4.4-cm, well circumscribed, and heterogeneously enhancing mass in the left adrenal gland (Fig. 1B). Evaluation of metastasis using positron emission tomography-CT was performed, and no definite metastasis was found. At this point, a provisional diagnosis of ACC or adenoma was made.
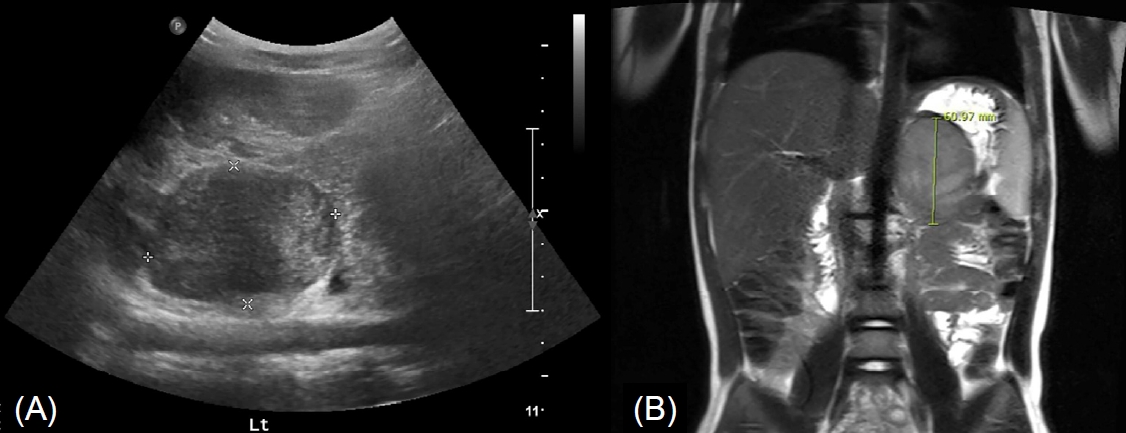
A well circumscribed, poorly heterogeneously enhancing mass in the left adrenal gland was noted on abdominal ultrasonogram (A) and magnetic resonance imaging (B).
After investigating metastasis, laparoscopic resection of the left adrenal gland with the tumor mass was performed. The resected mass was an encapsulated round mass measuring 6.0×5.5×4.4 cm in dimension and weighing 85.4 g (Fig. 2). The tumor cells in the specimen had low-grade nuclear atypia and low mitotic activity (1/10 high-power fields). Necrosis and capsular invasion were absent; however, venous invasion was present (>4 foci). A low-grade oncocytic variant of ACC was confirmed by pathologists. According to the Lin-Weiss-Bisceglia criteria, venous invasion of the oncocytic variant is a major criterion; therefore, the tumor was diagnosed as a malignant carcinoma [10]. Two weeks after resecting the adrenal tumor, the patient’s serum testosterone, DHEA-S, 17-OHP, and androstenedione levels decreased dramatically to within the normal range (Table 1). The patient was referred to a pediatric oncologist for postsurgical treatment. The patient did not undergo postoperative chemotherapy for recurrence since no metastatic features were found and was followed up by endocrinologists, oncologists, and pediatric surgeons.

Gross appearance of the resected mass.
To determine the genetic etiology of ACC, targeted next generation sequencing was performed to identify genetic defects. Therefore, a genetic counseling session was conducted with the parents. Informed consent was obtained from the patient's parents, and genetic analyses were conducted. The molecular analyses revealed a heterozygous form of c.1298_1299delinsG (NM_000244.3) that led to the p.(Pro433Argfs*17), a novel frameshift mutation of MEN1. The gene mutation was confirmed by Sanger sequencing and presumed to be a likely pathogenic variant according to the American College of Medical Genetics and Genomic (ACMG) guidelines [11]. This form of mutation has not been previously reported in the literature. Neither of the patient’s parents had the same gene mutation, suggesting that the patient’s mutation was a sporadic, de novo mutation in MEN1.
Discussion
In this case report, we presented a 3-year-old girl with an atypical case of precocious puberty with virilization and advanced bone age, with no breast development. Abdominal imaging revealed a functioning mass in the left adrenal gland, which was later confirmed to be ACC by biopsy. Signs of virilization disappeared within one month after resecting the tumor. Through genetic evaluation to determine the etiology of ACC in this pediatric patient, a genetic mutation in MEN1 was found, and it was a likely pathogenic variant. This mutation was not inherited from the patient's parents and was thus a de novo case.
Functioning ACC should be resected surgically, and complete resection is important for a favorable prognosis [12]. Owing to their highly malignant and invasive nature, many ACCs tend to recur within one year of tumor resection. Regular follow-ups with adrenal hormone tests and imaging studies for recurrence surveillance should be performed, usually every three months for the first year with interval increase thereafter [13]. When ACC recurs, repetitive surgical resection is performed if possible, and adjuvant chemotherapy is usually administered [14]. Previous studies have suggested the following favorable prognostic factors for ACC in children: age, <4 years; tumor size <10 cm; virilization alone; tumor volume <200 cm3; and early tumor stage with no metastasis [15,16]. Patients younger than 4 years show remarkably favorable prognosis and do not require post-surgical chemotherapy [15]. Fortunately, the present 3-year-old patient had features that were mostly favorable. Early diagnosis of ACC, before the age of 4 years, was crucial because ACC rapidly progresses and invades other organs. After complete excision of the mass, the patient did not undergo chemotherapy for remnant disease, and hormone levels were monitored regularly for surveillance of recurrence.
The gene mutation detected in this patient, however, indicated that ACC is not only a single sporadic disease but an abnormality of the endocrine physiology. Other endocrine diseases may manifest later in life. Regarding the clinical course of MEN1, this patient requires regular check-ups with a particular focus on the parathyroid gland, pituitary gland, pancreas, and other endocrine organs, including the adrenal gland. Surveillance of neoplasms in nonendocrine organs, especially in the gastrointestinal tract, should be maintained for the rest of the patient's life. According to a surveillance guideline for MEN1 patients and carriers published in 2019 [1], parathyroid function (serum calcium and parathyroid hormone) should be checked annually until the age of 8 years. Anterior pituitary function (prolactin and insulin-like growth factor) should be checked till until age 5 years, with MRI performed every 3 years. Generally, adrenal tumors are not in the surveillance category unless patients have a functioning tumor or tumor size larger than 1 cm. Therefore, the present patient should follow the surveillance guidelines for ACC.
During our search on PubMed.gov, we found only five cases of childhood ACC reported in Korea. Among them, 3 cases reported treatment modalities and prognoses of ACC [14,17,18]. Another was about virilizing ACC in Turner syndrome with a TP53 gene mutation, and the last was a case of virilizing adrenocortical oncocytoma with borderline malignancy potential based on the Lin-Weiss-Bisceglia criteria [19,20]. None of these previous cases had MEN1 mutation, which makes the present case the first report of childhood ACC with MEN1 mutation in Korea. In this case, MEN1 mutation was a frameshift mutation that was not previously reported in the population. According to the ACMG guidelines [11], this gene mutation is likely pathogenic for ACC in this child, although the exact penetrance and expressivity of the mutation need further investigation. Unlike other hereditary transmitted cases previously discussed, MEN1 mutation was sporadic in this case, and the finding was discordant with the well-known inheritance mode of MEN1, i.e., that MEN1 is usually inherited in an autosomal dominant manner. Further investigations of the patient's family may be needed to identify hereditary traits of MEN1.
In conclusion, early diagnosis of ACC in children is of great importance because intervention at an early stage leads to a favorable prognosis, thereby increasing the overall survival rate. Pediatric endocrinologists should collaborate with pathologists, surgeons, geneticists, and oncologists to efficiently manage these patients. This multidisciplinary approach could provide optimal results for children with ACC. Due to the MEN1 mutation, the patient may experience other endocrinologic problems in the future, which warrants regular follow-ups. Endocrinologists should keep track of hormonal surveillance before puberty, during puberty, and even after adolescence. With recent advancements in genetic evaluation tools, genetic etiologies of ACC can be readily found. These tools should be actively utilized for better understanding and proper management of childhood ACC. Future studies that discuss the clinical aspects of genetically errant ACC will provide more clues for understanding the pathophysiology of childhood ACC in clinical fields.
Ethical statement
Informed consent was obtained from the patient's parents, and genetic analyses were conducted.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: KSE, MBA; Data curation: KSE, MBA; Formal analysis: KSE, MBA, WKC; Methodology: KSE; Project administration: KSE; Visualization: KSE; Writing - original draft: KSE; Writing - review & editing: MBA, NYL, JY, JWL, MK, JHJ, MHJ, BS