Balanced assessment of growth disorders using clinical, endocrinological, and genetic approaches
Article information

Abstract
Determining the pathogenesis of pediatric growth disorders is often challenging. In many cases, no pathogenesis is identified, and a designation of idiopathic short stature is used. The investigation of short stature requires a combination of clinical, endocrinological, and genetic evaluation. The techniques used are described, with equal importance being given to each of the 3 approaches. Clinical skills are essential to elicit an accurate history, family pedigree, and symptoms of body system dysfunction. Endocrine assessment requires hormonal determination for the diagnosis of hormone deficiency and initiation of successful replacement therapy. Genetic analysis has added a new dimension to the investigation of short stature and now uses next-generation sequencing with a candidate gene approach to confirm probable recognizable monogenic disorders and exome sequencing for complex phenotypes of unknown origin. Using the 3 approaches of clinical, endocrine, and genetic probes with equal status in the hierarchy of investigational variables provides the clinician with the highest chance of identifying the correct causative pathogenetic mechanism in a child presenting with short stature of unknown origin.
Highlights
Pediatric growth disorders originate from a wide variety of etiologies, and in many cases no definite pathogenesis is ascertained, leading to the designation of idiopathic short stature. The clinician has the greatest chance of determining the correct etiology by using a balanced approach of clinical assessment, endocrinological evaluation, and genetic studies having equal status in the hierarchy of investigational variables.
Introduction
Growth disorders comprise the largest group of referrals to a pediatric endocrinology consultation service. It is well recognized that short stature can be the source of physical and emotional distress in childhood and that adult short stature resulting from late diagnosis, or inefficacy or nonresponsiveness to treatment, can result in psychological burden [1]. Although the psychological response to short stature in childhood is variable and ranges from clinical psychopathology to development of resilience [2], it is clear that early diagnosis of a disease process is of benefit to the individual and circumvents the complications of an unrecognized disorder [3].
The challenge of identifying the precise cause of short stature in individuals referred for a medical opinion should not be underestimated. In many cases, a causative pathogenesis is not ascertained, which is why the designation of idiopathic short stature (ISS) has become established in clinical practice and remains a label attached to many patients [4]. Linear growth is a sensitive marker of general health in childhood. There has been a realignment of the classification of growth disorders, which formerly emphasized the hormonal regulation of growth and its defects as being the primary cause of treatable growth disorders. It is now appreciated that the physiological process of growth plate chondrogenesis should be regarded as the gateway to successful height attainment. Consequently, defects in growth plate chondrogenesis are now classified as primary growth disorders [5,6]. Examples include skeletal dysplasias, dysmorphic syndromes (chromosomal disorders), and small for gestational age with failure of catch-up growth. Secondary growth failure results from a wide range of mechanisms which adversely affect chondrogenesis and include endocrine disorders such as growth hormone (GH) deficiency, chronic conditions such as malnutrition, celiac disease, Crohn disease, and renal disease, or physical factors such as radiation [4,6].
When a child with short stature and their parents present for a medical opinion, the pediatrician should take a broad view of the possible etiological origin of apparent growth failure [3]. A protocol for assessment is needed which incorporates both common and uncommon causes, allowing the pathogenesis of true pathological short stature to be identified and excluding overinvestigation of the child with a variant of normal growth, such as constitutional delay of growth and puberty (CDGP) and familial short stature (FSS), both of which have good adult height prognoses. Such a protocol was recently published by Wit et al. for use in the Netherlands [6].
Our aim in this review is to appraise the process of growth assessment and to recommend a format which provides a balance between essential clinical evaluation and the more sophisticated techniques of biochemical and genetic testing now available to the clinician. The primary aim is to achieve an early and precise diagnosis which instructs future management for the long-term benefit of the patient.
Assessment of the child with short stature
From the clinical perspective, the child referred with short stature statistically has a height more than 1.6 standard deviations (SDs) below the mean of the population or of the family, using the Hermanussen and Cole definition of target height [7]. In practical terms, the perception by the child or parents that height is below average is sufficient to warrant a specialist referral. Therefore, children referred to a pediatric endocrinologist can have highly variable degrees of 'short stature' or growth failure and equally wide ranges of potential etiologies, from complete normality to a growth pattern which will lead to adult height below the target range, that is, less than 1.6 SD below the midparental height [4].
Standard procedures for diagnostic assessment of short stature have been criticized for producing a low yield of positive diagnostic data. The general screening tests proposed in a consensus publication on guidelines for ISS evaluation [8] were criticized for low positive yield and lack of cost-efficacy [9]. Similarly, a low yield of diagnostic endocrine data was noted, with a recommendation that the traditional paradigm of growth assessment should move to a more genetic-centric model [10]. Our view is that investigational protocols should be frequently appraised and updated; yet in the case of a growth disorder, a balanced approach providing parity to the 3 key strands of clinical, endocrinological, and genetic evaluation is important and should be preserved. We will attempt to justify this approach below.
Diagnostic approach and investigation of patients with short stature
1. Clinical assessment
Algorithms for the clinical assessment of short stature patients have been published in a number of reviews [6,8,11]. A general scheme for clinical assessment of short stature is shown in Fig. 1. Clinical skills are crucial in eliciting a detailed family history, including consanguinity and the heights of parents, grandparents, and siblings, and in documenting a detailed and accurate pedigree for cases of possible genetic disease. Birth weight and length, gestation, and developmental milestones need to be documented, and potential symptoms from the major body systems need to be elicited by direct questioning. In a child born small for gestational age (SGA), a highly heterogeneous state, there are potential influences from geographical, maternal, paternal, placental, environmental, and fetal factors. The diagnostic approach to the short child should similarly start with a search to identify clues for a primary or secondary growth disorder. Primary growth disorders are clinically defined syndromes associated with growth failure such as Silver-Russell, Noonan, Prader-Willi, Bloom, 3M, and neurofibromatosis type 1 syndromes and skeletal dysplasias (growth plate chondrogenesis disorders) such as those caused by ACAN, FGFR3, NPR2, and SHOX gene mutations [12]. A careful medical history, including details of maternal lifestyle such as smoking and alcohol consumption and environmental conditions during pregnancy, is essential to elicit. Postnatal developmental delay, feeding difficulties, and behavioral problems are also important to probe for by direct questioning [4].

Scheme for clinical assessment of short stature.
Physical examination should include accurate auxology with measurement of height, sitting height, sitting height/height ratio (standard deviation score [SDS]), arm span, and head circumference [6]. Documentation of dysmorphic features which may be subtle, as in 3M syndrome and IGF1R variants, is an important skill that clinicians need to acquire. Body asymmetry and disproportion, microcephaly or relative macrocephaly, heart murmur, cryptorchidism, and muscular hypertrophy should be sought. Documentation of severe short stature (height < -3 SDS) and height SDS similar to one parent are also relevant clues for underlying genetic disorders [4,6].
2. Biochemical, endocrinological, and radiological investigations
1) General pediatric screening tests
The suggested scheme for laboratory screening tests to exclude general pediatric pathology, including thyroid function, insulin-like growth factor-1 (IGF-1), insulin-like growth factor binding protein-3 (IGFBP-3), and karyotype in girls, was proposed in the 2008 ISS Consensus Statement [8]. The practice of performing general pediatric tests was challenged in 2013 by Sisley et al. [9], who reported from the Cincinnati Children's Hospital that of 232 short patients who were healthy and had normal physical appearance, 98.7% had normal screening tests. The message in this publication was that screening tests were not cost-effective in healthy short children. To some extent this view was perpetuated in the recent Growth Hormone Research Society (GRS) 'Guidelines' article [13], which stated that 'Laboratory tests should be guided by clinical features rather than routinely applied to all patients with short stature ... Clinical discretion should be applied to the scope of testing for nonendocrine disease'.
We do not agree with this recommendation. In children with unexplained short stature, it is essential that routine hematological and biochemical screening tests are performed (Fig. 2). Chronic illnesses such as celiac and Crohn diseases need to be excluded, as do sex chromosome defects such as mosaic Turner syndrome [14]. These disorders cannot be excluded by clinical assessment alone. Short stature patients with nonFSS are most likely to have occult illness, being shorter than their parental target height, so pathological causes need to be excluded before endocrine assessment. Recombinant human growth hormone (rhGH) therapy may be prescribed in many children with ISS, and normal renal, liver, and hematological status are important to document before this therapy is initiated. Laboratory screening should consist of full blood count, erythrocyte sedimentation rate, IGF-1, free thyroxine, thyroid-stimulating hormone, a test for celiac disease, electrolyte and renal and liver function tests, calcium, phosphate, and alkaline phosphatase (Fig. 1) [6,8]. A hand/wrist bone age x-ray with examination for anatomical features of skeletal dysplasia should be performed in all referred short children. A karyotype or single nucleotide polymorphism array analysis (for detecting Turner syndrome, and with the latter tool also for copy number variants and uniparental disomy) in all girls with height < -2 SDS or height SDS < target height SDS -1.6 should also be performed [6]. If serum IGF-I is low, a GH stimulation test or an IGF-I generation test (IGFGT) are further options for identification of an endocrine etiology.
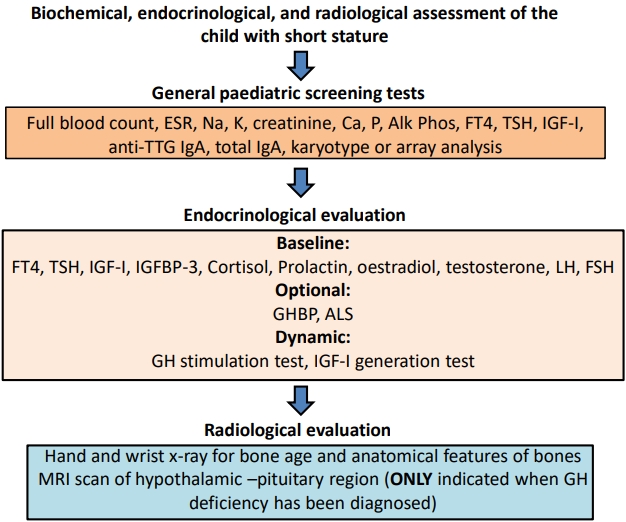
Biochemical, endocrinological, and radiological assessment of the child with short stature. ESR, erythrocyte sedimentation rate; Alk Phos, alkaline phosphatase; FT4, Free thyroxine; TSH, thyroid-stimulating hormone; IGF-1, insulin-like growth factor-I; TTG, tissue transglutaminase; IGFBP-3, insulin-like growth factor binding protein-3; GHBP, growth hormone binding protein; ALS, acid labile subunit; MRI, magnetic resonance imaging; GH, growth hormone.
2) Endocrinological investigation of the GH-IGF-I axis
In the 1980s and 1990s, the study of childhood linear growth focused on the function of different components of the GH–IGF-I axis, and enormous progress in the understanding of this axis was made [15]. The somatomedin hypothesis first published in 1957 [16] was revisited 50 years later [17] to show that the IGF system plays a key role in growth regulation, with both circulating and peripherally produced IGF-I having individual roles [18]. IGF-1 deficiency was reported to occur in a proportion of short patients with normal GH secretion [19], which placed some ISS patients in an intermediate position between GH deficiency and GH resistance, although some overlap existed.
Some ISS patients have a degree of functional GH insensitivity [20] with a broad range of generation of IGF-I in response to GH. The important 2007 study by Cohen et al. [21] reported that high doses of rhGH were needed to reach a serum IGF-I concentration of +2 SD. Evidence of subnormal generation of IGF-I was also demonstrated in studies by Selva et al. [22] and Buckway et al. [23] on responses in the IGFGT. Compared to normal subjects, ISS patients had basal IGF-1 levels below the normal mean, and after GH stimulation on days 5 and 8 of the IGFGT, IGF-I levels were significantly lower than normal, regardless of GH dose. These hormonal findings challenge the definition of ISS patients having no endocrine abnormality.
3) GH-IGF-I status
Endocrine assessment of GH secretion and the IGF system should follow the guidelines published previously [8] and reviewed in the recent GRS Consensus Statement [13]. Important messages are highlighted in this publication, including the guidance that IGF-I assays with reliable reference data be used alongside age-appropriate pediatric reference ranges and that serum IGF-I is influenced by many factors such as assay methodology, age, nutrition, and chronic illness [24]. An IGF-I level > 0 SDS at any age makes GH deficiency unlikely but not impossible [13.24]. The GH stimulation test (GHST) has been criticized over the years for being a nonphysiological stimulus and for lack of reproducibility. Precision of the timing of peak GH in certain tests such as the arginine and levodopa tests will increase their sensitivity [25].
The GHST remains of key importance in the assessment of short children with undefined etiology [4]. An abnormal GHST, with a peak GH level of <7 or <10 μg/L in the United States [13] and auxological features consistent with GHD, removes a patient from the 'ISS' category. A low IGF-I value without knowledge of GH status cannot distinguish GH deficiency from GH insensitivity [19]. Longitudinal observation and monitoring may lead to re-evaluation of the GH-IGF-I axis in some slowly growing children that may involve a repeat of the GHST. Once the distinction between GHD and non-GHD short stature has been made, further investigations to define etiology such as measurements of IGFBP-3, acid labile subunit, and an IGFGT, with its recognized disadvantages [26], are options.
3. Genetic investigations
We now move to the third component of short stature assessment, namely genetic analysis. A proposed scheme for genetic investigations is shown in Fig. 3. Advances in genetic techniques and particularly next-generation sequencing have resulted in increasing yields of positive genetic diagnoses in short children [12,27]. A range of genetic techniques are available, but those which can provide positive causative answers should be prioritized. Targeted evaluation of a single gene (candidate gene sequencing) is recommended for a child who shows characteristic clinical or laboratory features of a well-known genetic syndrome. However, in most cases there is no strong suspicion for a certain recognizable disorder, so the candidate gene approach has largely been overtaken by exome sequencing (ES), using targeted gene panels, supplemented by chromosomal microarray [4]. Gene defects which affect the endocrine control of growth will be discussed first, followed by defects of chondrogenesis affecting growth plate function.

Proposed scheme for genetic investigations of the child with short stature. SNP, single nucleotide polymorphism; CGH, comparative genomic hybridization; CNV, copy number variation; UPD, uniparental disomy.
1) Variants in genes regulating GH action in children with short stature
Short stature patients may have variable GH sensitivity and IGF-I concentrations [11]. Therefore, it has been suggested, but remains controversial, that fewer deleterious GHR gene defects may cause ISS associated with features of GH insensitivity [4]. Studies of ISS cohorts have reported heterozygous GHR variants occurring with a frequency ranging from 5% to 15.5% [28]. It has also been noted that although GHR sequence changes are common in children with ISS, many were also identified in control subjects and normal stature family members [29].
Since molecular investigations of short stature phenotypes started in the late 1980s, a number of pathogenic variants have been discovered in children labelled as having ISS or SGA. In 2019, Storr et al. [29] published an extensive review of mild or 'nonclassical' abnormalities of GH action. Mild forms of GH insensitivity can be divided into 3 categories: (1) aberrations of GH signaling caused by homozygous or heterozygous variants of genes encoding the GHR or signal transducer and activator of transcription 5B (STAT5B) [29-32]; (2) defects of IGF-I secretion (IGF1), transport (IGFALS), and bioavailability (PAPPA2) [29,33-35]; and (3) IGF-I insensitivity (IGF1R) [36]. Most patients with GH1, GHR, and STAT5B defects are born with a normal birth size, while patients with IGF1 and IGF1R variants are typically born with a low birth size. Paternally-transmitted heterozygous IGF2 variants are one of the recognized genetic causes of Silver-Russell syndrome [37]. A summary of phenotypic and endocrine features of genetic defects in patients with mild to moderate short stature is shown in Table 1.

Summary of phenotypic and biochemical features of defects causing growth hormone resistance originally labelled as ISS (4)
2) Gene variants affecting growth plate chondrogenesis
Genetic defects which impair chondrogenesis are likely to cause some degree of body disproportion; however, this may be mild and not noticed by the clinician, resulting in the child being labelled as ISS or SGA [38]. Out of the multiple reported genes [12], we discuss 4 examples.
(1) SHOX haploinsufficiency
Pathogenic defects of the gene encoding short stature homeobox (SHOX), located at the tip of the X and Y chromosomes, and deletions or duplications of the SHOX enhancer regions, impair chondrocyte differentiation in the growth plate. A gene dose effect is apparent as homozygous or compound heterozygous inactivating SHOX variants cause Langer mesomelic dysplasia, while heterozygous abnormalities cause a milder skeletal dysplasia, Leri–Weill dyschondrosteosis, with the classical Madelung deformity of the wrist, or present clinically as ISS. SHOX haploinsufficiency is caused more frequently by CNVs than by single-nucleotide variants [39] and is reported to account for 2%–15% of children presenting with ISS [40]. In a series of 521 patients with short stature due to SHOX haploinsufficiency treated under the license for treatment with rhGH, 44% were documented to have nonsyndromic short stature [41]. The SHOX haploinsufficiency phenotype is clearly broad and not yet well defined [42].
(2) Fibroblast growth factor signaling
Fibroblast growth factors (FGFs) and their receptors play a role in growth plate physiology. The FGF receptor 3, encoded by FGFR3, acts as a negative regulator of chondrogenesis, so heterozygous activating variants impair bone elongation resulting in short-limbed skeletal dysplasia [12]. There is a range of phenotypes, from classical achondroplasia to milder hypochondroplasia presenting as a relatively mild skeletal dysplasia, even with normal body proportions [43] and showing some response to rhGH therapy [44].
(3) CNP-NPR2 pathway
C-natriuretic peptide (CNP) encoded by NPPC is a local, positive regulator of growth plate function [12]. Homozygous inactivating variants of NPR2, which encodes the main CNP receptor, cause severe skeletal dysplasia, but relatives who are heterozygous for these mutations have a mild growth defect with phenotypes similar to SHOX deficiency [45]. Heterozygous NPR2 variants are thought to account for 2%–6% of cases of ISS [46,47].
(4) ACAN mutations
The growth plate is situated between the epiphysis and metaphysis of the long bones, and chondrogenesis proceeds with osteoblasts, osteoclasts, and blood vessels transforming the newly formed cartilage into bone. Aggrecan is the most abundant proteoglycan in hyaline cartilage and is crucial to the structure and function of the growth plate. Variants in ACAN, which encodes for aggrecan, are associated with a range of growth defects which may be severe or mild and which may present as ISS or SGA [12]. Patients carrying heterozygous variants of ACAN can reach an adult height of 150–152 cm without further dysmorphic features [48]. Hauer et al. [49] performed sequence analyses in 428 families with short stature, and the results showed that heterozygous nonsense variants of ACAN were identified in 6 families, that is, 1.4%. The mean height SDS value of the affected subjects was -3.2 SD, which is consistent with a label of ISS. In a study of WES of 200 short stature patients, Hauer et al. [50] also identified heterozygous carriers of recessive skeletal dysplasia alleles in 3.5% of cases. These were notably ACAN and NPR2 defects, with ACAN being the most commonly mutated known short stature-associated gene with a frequency of 2.5%.
3) Which children with short stature should have genetic investigations?
Clinically heterogeneous short children may harbor a range of molecular etiologies in up to 40% of cases [51-54]. Selection criteria for genetic testing and methods will continue to evolve. In general, genetic investigations are indicated if there are positive clinical diagnostic clues for a monogenic disorder. Examples of such clues include severe short stature, microcephaly or relative macrocephaly, dysmorphic features, disproportion, a positive family history, and a low birth size (SGA) [6]. In general, patients with a clinical suspicion of monogenic disorders in whom a clear diagnosis will enhance clinical management in terms of genetic counseling are good candidates for genetic testing. Examples include soft dysmorphic features suggestive of Noonan syndrome, patients with possible mild GH insensitivity [29], and patients with borderline body disproportion suggestive of SHOX, ACAN, or NPR2 genetic variants [5,55]. In general, syndromic rather than nonsyndromic cases are more likely to yield positive genetic results.
The principal aim of genetic analysis is to identify monogenic disorders having a significant effect on growth [10]. In children with ISS, one would expect that these defects would be found mainly in the non-FSS subjects, where parents with normal stature may be carriers of a recessive gene or the patient carries a de novo pathogenic gene variant. An example of apparent NFSS of genetic origin is the pseudoexon GHR variant, which can present with mild short stature and normal IGF-1 levels [56]. Many of the FSS patients are likely to have polygenic short stature, having inherited common gene variants of small effect size [51]. However, if dominant inheritance of short stature is traceable in one or more generations, monogenic defects as in NPR2 [51] and SHOX mutations, dominant negative GHR mutations, and Noonan syndrome defects, or an inherited CNV, are likely [4].
In short children born SGA, genetic causes appear to be more frequent than in ISS, although direct comparative studies have yet to be performed. Extreme examples include genetic defects in fundamental cellular processes, which can produce severe global growth deficiencies known as primordial dwarfism, where pre- and postnatal growth is severely affected. An extensive overview of gene variants associated with SGA was recently published by Finken et al. [38] As with ISS, the primary aim of genetic studies in SGA subjects is to identify monogenic defects. These may be predicted by the use of clinical scoring systems, such as the Netchine-Harbison system for Silver-Russell syndrome [57], the recently published scoring system for IGFR mutations [36] and concordance with characteristics of ACAN variants [55] or Madelung deformity, which is pathognomonic of SHOX haploinsufficiency. Positive scores for a recognized disorder can be followed up by candidate gene sequencing or, preferably using a hypothesis-free approach, with a genome-based targeted gene panel, including a check for copy number variations and uniparental disomy.
In a study of 55 unexplained cases of short SGA subjects, genotyping using a targeted gene panel or ES gave a positive diagnostic yield of 15% [58]. Heterozygous pathogenic or likely pathogenic genetic variants in 8 of the 55 patients were identified in genes already associated with growth disorders. Four of the genes were associated with growth plate development, IHH (n=2), NPR2 (n=2), SHOX (n=1), and ACAN (n=1), and 2 with the RAS/MAPK pathway, PTPN11 (n=1) and NF1 (n=1) [58]. In the case of a dysmorphic SGA child where the diagnosis was uncertain, ES was particularly helpful in identifying the mutation in the BLM gene confirming the diagnosis of Bloom syndrome [59]. In special cases where a novel monogenic disorder is suspected, ES in a 'trio,' that is, in the patient and both parents, can be performed. It is likely that future bioinformatic analyses of next-generation sequencing technology will show that in many cases, SGA is caused by a combination of multiple (epi)genetic variants [60].
We believe that a case can be made for the genetic assessment of short children born SGA, because the pretest likelihood of detecting a genetic condition is high (on the order of 30%–40%), particularly if a child presents with additional features as summarized previously. Another benefit of positive genetic analysis is to be sure that the child does not have a syndrome for which rhGH treatment is contraindicated (e.g., Bloom syndrome) or debatable (e.g., neurofibromatosis type 1) [61]. A third reason is that the positive diagnosis of IGF1R haploinsufficiency or genetic syndromes affecting growth plate function (e.g., SHOX haploinsufficiency) implies that a higher dosage of rhGH is indicated to generate effective growth acceleration.
4) Which short patients should NOT have genetic investigations?
The decision about genetic testing in a short child must be made by the clinician responsible for the child's care by subjectively weighing the diagnostic clues for a primary growth disorder, their severity, and potential impact on management [12]. It is reasonable to assume that when the label of ISS or SGA is given to a child, a detailed auxological examination would have been performed to exclude body disproportion and major dysmorphic features. Similarly, endocrine investigations to exclude GH deficiency and GH insensitivity should have been performed. In general, the greater the severity of short stature, the more likely it is that there is an identifiable genetic defect [13], but the number and severity of any additional congenital anomalies or dysmorphic features and evidence of a skeletal dysplasia, associated intellectual disability, microcephaly, or relative macrocephaly should be added to the interpretation [6,10,51]. Not all short children should have genetic investigations [13]. Specifically, children with mild FSS without any additional abnormal clinical features should not be tested. Similarly, children with delayed bone age with expected adult heights within the normal range with a positive family history for delayed puberty for the family should in general not be tested as they may have CDGP or polygenic FSS.
Conclusion
The etiological possibilities for short stature are so broad and numerous that identification of a precise pathogenesis in a short child will always remain a challenge. Clearly, the arrival of next-generation genetic sequencing has added a new dimension to the investigational process. However, we do not believe that clinical skills should be marginalized in favor of high-technology genetic analysis. There is a danger that the clinician will be drawn increasingly to look at the computer screen, rather than engage with the child and family. The clinical approach to the patient and family is of prime importance for 3 key reasons: first, the establishment of the patient-physician relationship [62]; secondly, eliciting an accurate history, both to establish a detailed pedigree and to probe for symptoms of body system dysfunction by direct questioning; and thirdly, for the careful documentation of the phenotype. Endocrine assessment will arguably provide the least precise information; however, hormone determination is necessary if effective replacement therapy is to be used. For these reasons, we advocate that clinical, endocrine, and genetic assessments have equal status in the hierarchy of investigational variables [4]. Such an approach can fulfill the requirements of precision medicine and lead to a comprehensive approach to short stature evaluation with positive diagnostic results.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.