Low-dose mitotane-induced neurological and endocrinological complication in a 5-year-old girl with adrenocortical carcinoma
Article information

Abstract
Mitotane is an adrenolytic drug that exhibits therapeutic effects within a narrow target range (14–20 μg/dL). Various complications develop if the upper limit is exceeded. We present the case of a 5-year-old girl with breast development, acne, and pubic hair who was diagnosed with an adrenal mass that was subsequently excised. The pathological finding was adrenocortical carcinoma with a high risk of malignancy, and adjuvant therapy (combined mitotane and radiation therapy) was recommended. Mitotane was initiated at a low dose to allow monitoring of the therapeutic drug level, and high-dose hydrocortisone was also administered. However, the patient exhibited elevated adrenocorticotropic hormone levels and vague symptoms such as general weakness and difficulty concentrating. It was important to determine if these symptoms were signs of the neurological complications that develop when mitotane level is elevated. Encephalopathy progression and pubertal signs appeared 6 months after diagnosis, induced by high mitotane level. The mitotane decreased to subtherapeutic level several months after its discontinuation, at which time endocrinopathy (central hypothyroidism, hypercholesterolemia, and secondary central precocious puberty) developed. The case shows that low-dose mitotane can trigger neurological and endocrinological complications in a pediatric patient, indicating that the drug dose should be individualized with frequent monitoring of the therapeutic level.
Highlights
· The major difficulty in mitotane treatment is related to a narrow therapeutic window.
· Even low-dose regimen can induce complications if the drug levels exceeds the target range.
· The dose should be individualized through frequent monitoring to optimize treatment and minimize complications.
Introduction
Mitotane is an adrenolytic drug and is the only approved agent for treatment of adrenocortical carcinoma (ACC) [1], a rare neoplasm characterized by a poor prognosis with a 3- to 4-year median overall survival [2]. Adjuvant mitotane treatment of adults reduces postoperative recurrence by 38% and the postoperative death rate by 31% [3]. The major goal of mitotane treatment is to maintain the plasma level above 14 mg/dL to ensure an antineoplastic effect but below 20 mg/L (the upper limit of the therapeutic window) to avoid complications, especially neurotoxicity [4]. Two strategies inform the starting dose and escalation period in adults. Mitotane is initiated at a low dose (1 g/day with progressive weekly increases up to 2–3 g/day) [5] or a high dose (3 g/day rapidly increasing to 6–9 g/day within 2 weeks) [6]. However, few data are available on the appropriate drug dose, effectiveness, or frequency of complications in pediatric patients with ACC [7-9]. To the best of our knowledge, only one case has been reported, where therapy-related encephalopathy developed after prescription of high-dose mitotane (5 g/day) [10].
We present the case of a 5-year-old girl who developed neurological and endocrinological complications during low-dose mitotane treatment for ACC. We also briefly review the mechanisms of mitotane-induced complications.
Case report
A 5-year-old girl was admitted to the Dong-A University Hospital with breast development, acne, and pubic hair. Breast budding and acne had commenced 1.6 years prior (at age 3.6 years), and she had visited the outpatient clinic 6 months prior (at age 4.6 years). She exhibited a prepubertal response (a peak luteinizing hormone level of 1.38 mIU/mL in the gonadotropin-releasing hormone [GnRH] stimulation test) and a high serum basal estradiol level (74.32 pg/mL), suggestive of peripheral precocious puberty. Although ultrasonography revealed no abnormal pelvic lesions, breast development and acne progressed and pubic hair had begun to appear 5 months prior (at 4.7 years of age). The patient was born at a gestational age of 38 weeks (birth weight 3.2 kg). There was no familial medical history of note (no tumor and no adrenal gland, thyroid, lipid metabolism, or neurological disease).
At admission (at age 5 years), the patient's height was 109.9 cm (50th–75th percentile), her weight 21.3 kg (75th–90th percentile), and her body mass index 17.6 (90th–95th percentile). Her blood pressure was 110/66 mmHg. Growth was accelerated (10.4 cm/yr), and Tanner stage III breasts, stage II pubic hair, and facial acne were observed. The serum concentrations of estradiol, dehydroepiandrosterone-sulfate (DHEA-S), and 17-hydroxyprogesterone were elevated (Table 1). Her bone age (BA) was advanced (11.0 years). Computed tomography confirmed a large hypervascular mass in the left suprarenal area with no metastatic lesion, consistent with ACC (Fig. 1A). The patient underwent laparoscopic left adrenalectomy, the pathology of which was diagnostic of ACC. The tumor weighed 198 g and was 8.5×7.0×5.8 cm in dimension with a Ki-67 protein proliferation index of 10%. The tumor was classified as stage II (pT2 N0 M0) [11]. associated with a high risk of malignancy (a score of 3 using the Wieneke Index criteria; a score of 7 in the modified Weiss system) (Fig. 1B) [9]. Targeted next-generation sequencing (CancerSCAN level 2 panel, Customized SureSelect Targeted Exome Kit [Agilent], Illumina NextSeq 550Dx, LabGenomics) of TP53, MENIN, MSH2, MSH6, MLH1, PMS2, IGF2, APC, NF1, and PRKAR1A was performed on formalin-fixed paraffin-embedded adrenal tissue but revealed no pathogenic variant. No deletion or duplication was identified on TP53 multiplex ligation-dependent probe amplification analysis of peripheral blood.

Changes in Tanner stage, bone age, and hormonal laboratory findings after diagnosis
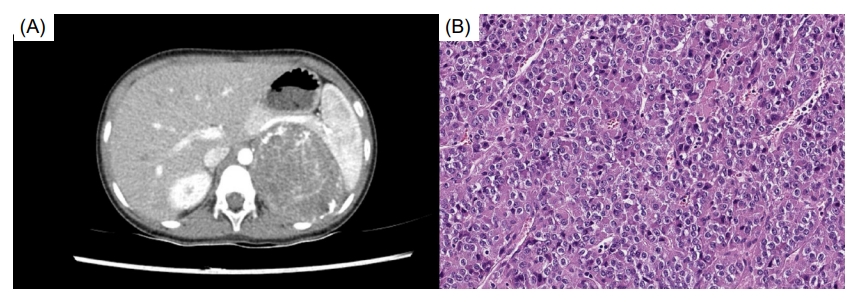
(A) Computed tomography revealed a large hypervascular mass (7.3 × 7.0 cm) in the left suprarenal area, consistent with adrenocortical carcinoma. (B) Histopathological analysis of the adrenalectomy specimen revealed high nuclear grade and mitotic figures (H&E, x400).
The patient was administered adjuvant therapy (a combination of mitotane and radiation therapy to the left abdomen [45 Gy in 30 fractions]). Mitotane was initiated at a low dose (1.25 g/m2/day) with monitoring of the serum level (Fig. 2). High-dose hydrocortisone (30 mg/m2/day) was also prescribed. During the first 2 months after diagnosis, the patient developed general weakness, epigastric discomfort, cognitive and concentration disturbances, and behavioral regression. A psychiatric consultation was requested because of possible acute stress disorder and/or attention-deficit hyperactivity disorder. The serum level of mitotane was assayed approximately 1 month after initial testing. Both the first and second levels were subtherapeutic (<0.4 and 7.8 μg/dL), and the drug dose was gradually increased to 3.0 mg/m2/day. The adrenocorticotropic hormone (ACTH) level remained within the normal range (16 pg/mL).
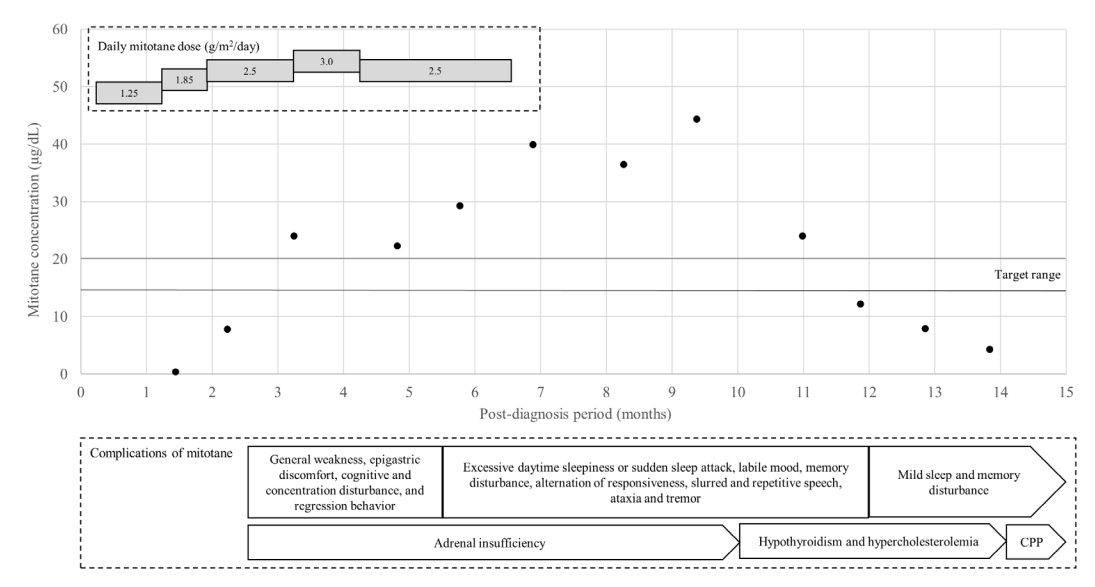
Serum concentration of mitotane and the daily doses and complications over time. Neurological complications began to appear when the mitotane serum level exceeded the target. The drug level gradually increased despite reduction of the dose and required 5 months to fall to within the target range after discontinuation. The numbers in and the lengths of the grey rectangles represent the daily doses relative to the body surface area and the durations of such drug administrations, respectively. The contents of the white rectangle are the mitotane complications that occurred during that period. The contents of the pentagon are the complications that persisted. The black dots are the measured mitotane serum concentrations, while the black lines represent the target range (14–20 μg/dL). CPP, central precocious puberty.
Three months after diagnosis (at age 5.3 years), her breast development had regressed to Tanner stage II, the facial acne had disappeared, and the serum DHEA-S level had decreased to within the normal range (Table 1). The mitotane level was elevated to 24.0 μg/dL, with persistent neurological symptoms, and the dose was promptly decreased to 2.5 mg/m2/day (Fig. 2). The hydrocortisone dose was adjusted to 54.8 mg/m2/day to normalize the ACTH level.
Six months after diagnosis (at age 5.5 years), the patient was re-admitted because of progression of the pubertal signs (breast Tanner stage II–III). The serum estradiol and sex hormone binding protein levels were markedly increased (Table 1), and pelvic ultrasonography revealed a thickened endometrium and an enlarged uterus with more advanced BA. However, computed tomography and 18F fluorodeoxyglucose positron emission tomography revealed no recurrence. The patient also developed excessive daytime sleepiness, sudden sleep attacks, memory disturbances, changes in responsiveness, slurred and repetitive speech, a labile mood, ataxia, and tremors. However, pituitary magnetic resonance imaging and autoimmune and cerebrospinal fluid analyses revealed no abnormalities. An electroencephalogram showed continuous generalized fast activities possibly associated with encephalopathy. Given the pubertal progression, the possibility of a hidden malignancy could not be excluded, and mitotane treatment was continued at a slightly decreased level (2.5 g/m2/day). However, her neurological symptoms worsened. The drug level had increased to 29.3 μg/dL, and mitotane was discontinued (Fig. 2). An aromatase inhibitor was initiated to treat the progression of pubertal signs because the patient exhibited a prepubertal response on the GnRH stimulation test (Fig. 3, Table 1).
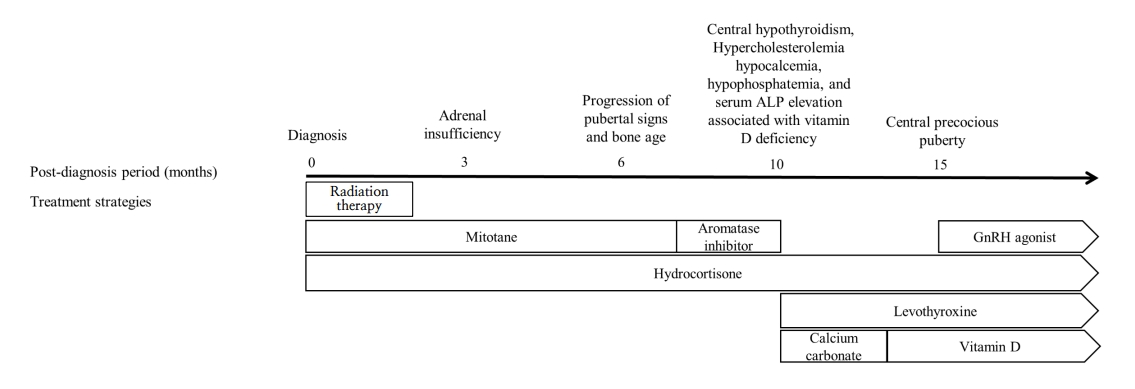
Treatment by time after diagnosis. Hydrocortisone, levothyroxine, and a gonadotropin (GnRH) agonist were used to treat adrenal insufficiency, hypothyroidism, and central precocious puberty, respectively (complications of mitotane use). Calcium carbonate and vitamin D were employed to treat hypocalcemia caused by vitamin D deficiency, hypophosphatemia, and elevated alkaline phosphatase (ALP) level. The numbers above the black arrow are the times (months) after diagnosis. The white rectangle contains the treatments used during that period. The contents of the pentagon are the maintained treatment strategies.
Ten months after diagnosis (at age 5.8 years), the patient was referred to the Seoul National University Children’s Hospital to monitor her symptoms. Her Tanner stages were breast III and pubic hair II, and her BA had advanced to 12.0 years. Laboratory tests revealed a high ACTH level, central hypothyroidism (free thyroxine, 0.53 ng/dL; thyroid-stimulating hormone, 3.31 μIU/mL), hypercholesterolemia (total cholesterol, 229 mg/dL; triglycerides 161 mg/dL; high-density lipoprotein cholesterol, 70 mg/dL; low-density lipoprotein cholesterol, 148 mg/dL), hypocalcemia (8.4 mg/dL), hypophosphatemia (3.3 mg/dL), and a high serum level of alkaline phosphatase (814 IU/L) associated with vitamin D deficiency (12.1 ng/mL). Repeat autoimmune and cerebrospinal fluid analyses revealed no abnormalities. An electroencephalogram revealed similar findings of continuous generalized fast activities with minimal variability and reactivity, possibly indicative of diffuse encephalopathy. The patient was maintained on hydrocortisone, and levothyroxine, calcium carbonate, and vitamin D were initiated (Fig. 3). The serum level of mitotane was above the target level for 3 months after discontinuation (up to 44.4 ug/dL), but the neurological manifestations improved as the mitotane level decreased below the subtherapeutic level to 12.2, 7.9, and 4.3 μg/dL (Fig. 2).
Fifteen months after diagnosis (at age 6.3 years), the mitotane level had decreased, but the patient’s breast development progressed again and BA became more advanced. The GnRH stimulation test revealed central precocious puberty, and a GnRH agonist was initiated. At the last follow-up (at age 6.6 years), she continued to be administered hydrocortisone, levothyroxine, and a monthly GnRH agonist without evidence of tumor recurrence and exhibited only mild sleep and memory disturbances.
Discussion
ACC is a rare endocrine tumor with an overall annual incidence of approximately 0.2–0.3 new cases per 1 million children [9]. The prognosis is generally poor, with a 5-year overall survival rate in children ranging from 30% to 90% depending on age and tumor stage [7,9]. Surgical resection is the principal strategy for operable ACC, but patients with advanced ACC (which may be recurrent, metastatic, or inoperable) may benefit from adjuvant therapy [11].
Mitotane has been used to treat advanced ACC since 1959. The drug destroys the inner zones of the adrenal cortex (zona fasciculata and reticularis) and exhibits tumor specificity because the adrenolytic effects seem to be enhanced by the cytochrome P450 (CYP) 11B activities of tumors [12]. The mechanism of action is not completely understood, but a previous study using ACC cells established an effect of mitotane on the mitochondrial respiratory chain by inducing a defect in cytochrome C oxidase activity and inhibiting sterol-O-acyl transferase 1 to trigger endoplasmic reticulum stress and apoptosis [13]. In addition, mitotane decreases the levels of messenger ribonucleic acids encoding CYP11A1 and CYP17A1, which are involved in cortisol and DHEA-S biosyntheses in the adrenal gland [14].
Given its highly lipophilic nature, 35-40% of mitotane absorbed from the gastrointestinal tract is distributed within adipose tissue [15]. In adults receiving a low-dose regimen, 3 to 4 months are required to attain the target serum range (14–20 μg/dL) [5]. The high-dose regimen reduces the time required to attain the therapeutic level at the cost of lower tolerability and greater toxicity [6]. No consensus exists on mitotane use in pediatric patients, and the drug dose is adjusted based on body surface area just as in adults [8,9].
Our patient was placed on low-dose mitotane with careful dose escalation accompanied by monthly monitoring of the drug level. Although the therapeutic level was achieved at 2 months, an elevated level was detected at the time of the next monitoring, perhaps reflecting drug accumulation. The drug level remained elevated despite prompt dose reduction. Several months elapsed before the level fell to within the target range as mitotane has a very long half-life (18 to 159 days). During that period, the patient exhibited mitotane-induced encephalopathy, the severity of which reflected the drug level [16]. Moreover, at least 1 month is required for assay of mitotane level after initiation. Thus, even low-dose mitotane can increase the body level above the therapeutic range. Drug commencement, escalation, and monitoring should be individualized with frequent testing, especially in pediatric patients with ACC.
Mitotane not only inhibits adrenal gland steroidogenesis, but it also induces liver CYP3A4 activity, increasing cortisol catabolism [11]. Hydrocortisone at a supraphysiological dose is always recommended when commencing mitotane [11]. Mitotane increases the level of cortisol-binding protein, artificially raising the total cortisol level [11]. Neither 24-hour urine excretion nor serum cortisol level is a reliable measure of adrenal insufficiency, while the ACTH level is a pulsatile indirect parameter. However, despite maintenance of high-dose hydrocortisone therapy, the ACTH level was elevated and accompanied by vague symptoms such as general weakness and concentration difficulties. The patient's ACTH level remained high even 5 months after mitotane discontinuation, hindering management of her various symptoms. Therefore, it is important to determine whether symptoms are an early sign of neurologic complications caused by mitotane, even if adrenal insufficiency is apparent. In addition, maintenance of high-dose hydrocortisone accompanied by clinical and biochemical evaluations of adrenal insufficiency and mitotane level monitoring are important to identify the cause of vague symptoms.
Mitotane exhibits weak estrogen-like activity, with a binding affinity for the human estrogen receptor 1,000-fold lower than that of 17β-estradiol [17]. Progression of pubertal signs and an elevated estradiol level were observed during mitotane treatment of our patient, without any evidence of tumor recurrence. Mitotane elevates the sex hormone binding globulin level [11], which may have contributed to the observed increase in estradiol. Male patients can develop gynecomastia caused by both an estrogenic effect and strong inhibition of 5α-reductase activity by mitotane [11]. Our patient also exhibited several endocrinological complications, including hypothyroidism, hypercholesterolemia, and secondary central precocious puberty. Mitotane inhibits both the expression and secretion of thyroid-stimulating hormone and blocks the response to thyrotropin-releasing hormone [18]. Mitotane impairs deiodinase activity, triggering a marked reduction in free thyroxine level [17]. These effects cause biochemical features consistent with central hypothyroidism. Hypercholesterolemia (increased levels of both low-density lipoprotein- and high-density lipoprotein cholesterol) reflects both the reduced steroidogenesis (from cholesterol to pregnenolone) and the estrogenic effect [17]. Finally, early-life exposure to sex steroids and endocrine disruptors may affect both pubertal onset and tempo [19], triggering secondary central precocious puberty.
In conclusion, low-dose mitotane, which is thought to be safer than the high-dose regimen in adults, can cause neurological and endocrinological complications in pediatric patients. Although these are reversible, the recovery time is long. The long-term effects on brain function, development, and growth remain unknown. Therefore, the drug dose must be individualized in pediatric patients, and it is important to recognize the early signs of complications and to frequently monitor the drug level to optimize the therapeutic effect and minimize complications.
Ethical statement
This case was approved by the Institutional Review Board of Seoul National University Hospital (approval number: 2102-052-1196). Informed consent was obtained from the patient and her parents.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: JHY, YAL; Data curation: YJH; Project administration: YSC, SHP, SBL, HAK, JYC, BCL; Writing - original draft: YJH; Writing - review & editing: YAL, BCL, HWC