Genotype - phenotype correlation in an adolescent girl with pathogenic PPARy genetic variation that caused severe hypertriglyceridemia and early onset type 2 diabetes
Article information

Abstract
Severe hypertriglyceridemia (HTG) (>885 mg/dL) can be caused by familial partial lipodystrophy type 3 (FPLD3), an autosomal dominant disorder caused by loss of function of the peroxisome proliferator-activated receptor gamma (PPARG), characterized by abnormal distribution of fat and metabolic derangements. This case reports a 16-year-old female (body mass index, 23.5 kg/m2) hospitalized twice for pancreatitis (triglycerides [TG] level >2,200 mg/dL). Her treatment management included bowel rest, insulin infusion, and plasmapheresis. A low-fat diet with 10 g of fat daily and 160 mg of fenofibrate daily decreased fasting TG to 411 mg/dL (range, 0–149 mg/dL). The patient had a normal leptin level. Panel testing of genes that impact TG metabolism revealed a known pathogenic variant in the PPARG gene (c.452A>G p.Tyr151Cys). A second variant detected in this gene, c.1003G>C (p.Val335Leu), is considered benign. Her glycosylated hemoglobin of 6.6% and 2-hour oral glucose tolerance test confirmed type 2 diabetes mellitus (T2DM). This study reports the earliest detection of T2DM in an adolescent with a pathogenic variant of PPARG. PPARG-related FPLD3 should be considered in lean children that present with severe HTG and insulin resistance, and subsequent treatment with proliferator-activated receptor gamma agonists, specifically thiazolidinediones, should be considered.
Highlights
· Severe hypertriglyceridemia (HTG) can be caused by familial partial lipodystrophies (FPLD).
· PPARG gene mutation causes FPLD type 3 characterized by abnormal fat distribution and severe HTG.
· Type 2 diabetes can occur early, in their teens, in a patients PPARG related mutation.
Introduction
Familial partial lipodystrophy (FPLD) is a heterogeneous genetic disorder that affects an estimated one in one million people and is characterized by abnormal regional distribution of adipose tissue. Six types of FPLD have been described: FPLD type 1 (Kobberling lipodystrophy); FPLD type 2, the most common type, is a defect in the lamin A/C (LMNA) gene (Dunnigan variety) [1]; FPLD type 3 causes a defect in the peroxisome proliferator-activated receptor gamma gene (PPARG-FPLD3) [2]; FPLD type 4 is related to pathogenic variants in the PLIN1 gene that codes for perilipin 1 [3]; FPLD type 5 is autosomal recessive and related to pathogenic variants in the CIDEC gene; and FPLD type 6 is caused by the LIPE (lipase E, hormone sensitive type) gene. While loss of adipose tissue (lipoatrophy) is the hallmark characteristic, clinical features include cutaneous (restrictive dermatopathy and alopecia) and skeletal anomalies (late closure of cranial sutures, bird-like progeroid facies). Metabolic aberrations include insulin resistance, diabetes, hypertriglyceridemia (HTG), low high-density lipoprotein (HDL) cholesterol, hepatic steatosis, and increased risk for cardiovascular disease. In most studies describing the metabolic aberrations in FPLD, HTG usually precedes glucose abnormalities, though insulin resistance is universal. FPLD can be clinically diagnosed based on loss of adipose tissue and documentation of abnormal fat distribution using whole body magnetic resonance imaging.
Peroxis ome proliferator-activated receptor gamma (PPARy) is a nuclear hormone receptor in the superfamily of ligand activated transcription factors (α, β /δ and У). PPARy is a critical key regulator of adipogenesis, the process in which mesenchymal stem cells change morpholog y, undergo clonal expansion, and insulin receptors are formed in mature adipocytes [4,5]. PPARy forms a heterodimer with retinoid X receptors, enabling its binding to the target to induce differentiation of preadipocytes to adipocytes, and it regulates several transcription factors that control adipocyte differentiation, resulting in small insulin sensitive adipocytes. It also upregulates adiponectin to improve insulin sensitivity [5]. The first description of heterozygous, dominant loss-of-function human mutations of PPARy was in 1999, and research has indicated that loss of PPARy function impacts both adipocyte differentiation and function [2].
Case report
A 16-year-old Hispanic female was hospitalized and received intensive care due to 2 pancreatitis episodes that occurred 1.5 months apart. On her first admission, her weight was 65.2 kg (82%, z-score=0.93), height 163.4 cm (54%, z-score=0.10) and body mass index (BMI) was 23.5 kg/m2. She presented with a 2-day history of abdominal pain, emesis, and anorexia. Computed tomography (CT) scans showed no appendicitis, extensive peripancreatic fluid with extension into the right pararenal space, and paracolic gutter. She was afebrile but tachycardic up to the 120s. Table 1 shows laboratory results at baseline, during admission, and after discharge. On admission, lipase was mildly elevated at 113 U/L (reference range, 8–78 U/L) and amylase was 123 U/L (25–125 U/L), which was at the high end of normal. White blood cell (WBC) count was 19,000/μL (range, 4,500–11,000/μL), hemoglobin (Hb) 13.5 g/dL (2–16 g/dL), hematocrit (Hct) 37.1% (36%–46%) with a normal coagulation profile (prothrombin time, 10.5 seconds; international normalized ratio, 1.02). Urine analysis results were 1+ ketones, 2+ bilirubin, 3+ protein, 1+ nitrites, 1+ leukocyte esterase, and 2–5 WBCs. Her sodium was 132 mmol/L, with mild lactic acidosis (bicarbonate of 21 mmol/L and lactate of 4.5 mmol/L). Her liver function test results were unremarkable. Eight hours later, her lipase and amylase normalized to 70 and 88 U/L, respectively. After 18 hours off of oral feeding and intravenous fluids, her triglycerides (TG) were reported as 1,920 mg/dL with a total cholesterol (TC) of 43 mg/dL with low HDL of 30 mg/dL. One day later, her TG rose to >2,200 mg/dL, TC of 426 mg/dL, and HDL of 34 mg/dL. Abdominal ultrasound revealed hepatomegaly and steatosis. No gall stones were identified. She was discharged 3 days later with recommendations of a low-fat diet and follow-up with a gastroenterologist after symptomatic improvement when she was able to tolerate oral feeding.
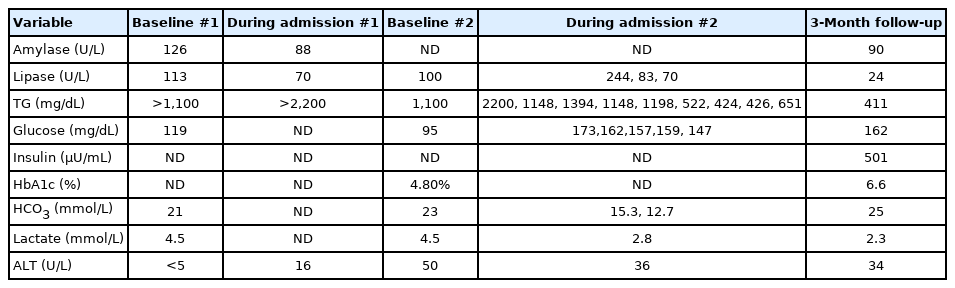
Laboratory results on baseline and during admissions (1 and 2) and follow-up appointment
She presented for her second admission 42 days later with recurrent pancreatitis. She had a pulse of 108 beats per minute and blood pressure (BP) of 144/70 mmHg. She was started on pain medications, IV ciprofloxacin, metronidazole, hydration, and bowel rest. On admission, her lipase was 3 times higher than normal, amylase was normal, TG were >1,100 mg/dL and increased to >2,200 mg/dL, TC was 1,200 mg/dL, and HDL was >5 mg/dL. Laboratory test results indicated a 17,000/μL, Hb 12 g/L, and Hct 33.4%. Erythrocyte sedimentation rate was 20 mm/hr. Thyroid function tests were normal, and glycosylated hemoglobin (HbA1c) was 4.8%. Random blood sugar measurements during her admission were elevated and ranged between 137–173 mg/dL. Elevated BP was attributed to pain, fluid overload, and hypernatremia and was treated with diuretics. A CT scan showed edema of the pancreas with no necrosis/calcifications. Insulin drip was started at 0.1 U/kg/hr. Three days into admission, plasmapheresis was performed and then repeated twice (30 and 50 hours after her first round) to assess worsening symptoms of epigastric pain, tachycardia, and metabolic acidosis (PH, 7.25; PCO2 mmHg, 29; PO2 mmHg, 44; HCO3-meq/L, 12.7). After 10 days, the patient was discharged on a strict low-fat diet of 10 g of fat daily, omega 3 acid ethyl esters (brand name Lovaza, 2 g), and fenofibrate 160 mg daily, which she tolerated well with no reported side effects.
Secondary causes including obesity, metabolic syndrome, and medication effects (e.g., glucocorticoids, anabolic steroids, retinoids, and diuretics) for HTG were ruled out.
Panel testing of genes involved in lipid metabolism was assayed on an Illumina MiSeq platform and VarSeq CNV (GRCh37) through the Blackburn Cardiovascular Genetics Laboratory at the Roberts Research Institute. This panel includes genes involved in lipoprotein metabolism: LPL, APOC2, APOA5, APOE, APOC3, LMF1, and GP1HBP1; intracellular carbohydrate metabolism: GCKR and GPD1; intracellular lipid metabolism: CREB3L3; as well as PPARG, a regulator of adipocyte differentiation. Results of LPL, APOC2, APOA5, LMF1, GPIHBP1, GCKR, CREB3L3, GPD1, APOE, and APOC3 gene analyses were normal. Two variants, c.452A>G (p.Tyr151Cys) and c.1003G>C (p.Val335Leu), were reported in the PPARG gene. The c.452A>G (transcript reference NM_015869.4, hg37 reference) p.Tyr151Cys variant in exon 3 was previously reported as pathogenic in the Human Gene mutation database [6-8]. The second variant, c.1003G>C (p.Val335Leu), is rare (minor allele frequency 0.01%). Current interpretations of this mutation in ClinVar are conflicting and assessments include "variant of uncertain significance," "likely benign," and "benign," as defined by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [9]. A family history was collected, and the following were noted: the father was 90 kg and denied hypertension and diabetes; the mother lived in the Dominican Republic and a had history of abnormal cholesterol, although no test results were available for review. The patient had not lived with her mother since 6 years of age. No significant family history was determined from other family members on either side of the family with regard to body fat distribution, diabetes, or lipid profile. Because the parents refused genetic testing, we could not ascertain whether the mutation was de novo versus an inherited variant.
A more detailed physical exam was conducted after the patient's genetic test results were received; based on her history, the fat loss in the distal part of her upper and lower extremities and slight excess of subcutaneous fat in the face and neck were consistent with lipodystrophy (family refused photographs and no skin fold thickness measurements were performed). There was no evidence of hirsutism. She had mild acanthosis nigricans on her neck and axillae. No eruptive xanthomas, xanthelasma, or lipemia retinalis were noted on exam. The patient had regular menstrual periods with no clinical concerns for polycystic ovaries. After 4 weeks of dietary fat restriction with fenofibarte and Lovaza, her fasting TG were 411 mg/dL, HDL, 28 mg/dL normal [N] >39 mg/dL), low-density lipoprotein, 84 mg/dL (N <100 mg/dL), TC, 151 mg/dL (N <170 mg/dL), and creatinine kinase was 90 IU/L (N, 29–168 U/L). Follicle stimulating hormone was 5 mIU/mL (N, 3–17 follicular phase), luteinizing hormone was 2.0 mIU /mL (N, 8–89 follicular phase), estradiol was 14.9 pg/mL (N, 2–266), total testosterone was 5.0 ng/dL (N, 9–58), and dehydroepiandrosterone sulfate 142 μg/dL (N, 63–373), which excluded polycystic ovary syndrome. She had a normal leptin level of 1.6 ng/mL (no established reference for adolescents). To evaluate the effect of the identified pathogenic variant in PPARG on glucose metabolism, as well as a history of elevated random sugars during her second admission, a 2-hour oral glucose tolerance test was performed using 75-g oral glucose. Her fasting glucose was 99 mg/dL, and her 2-hour postprandial glucose was 220 mg/dL. She had hyperinsulinemia: her insulin was 52 and 123 μU/mL at fasting and at 2 hours, respectively. Her HbA1c was 6.6%. These results confirmed type 2 diabetes mellitus (T2DM), and we recommended that she begin taking the PPARy agonist pioglitazone.
Discussion
Our patient was found to harbor a previously reported pathogenic variant in the PPARG gene, a rare cause of HTG. This diagnosis was made possible because of the genetic testing performed at the Blackburn Cardiovascular Genetics Laboratory, which exceeded the testing for the 5 known genes that cause familial homozygous HTG (LPL, APOC2, APOA5, LMF1, GPIHBP1). Broekema et al. [10] described a total of 97 FPLD3 associated PPARG variants, of which 14 children and adolescents presented with clinical phenotypic loss of adipose tissue, HTG and/or pancreatitis, and noted that symptoms usually presented around puberty, although lipoatrophy was described in an infant (compound heterozygote) and a 3-year-old male toddler. Majithia et al. [11] described 3 females with PPARG pathogenic variants with impaired glucose tolerance and/or type 2 diabetes (Table 2 compares the metabolic profiles of these patients with our patient).

Clinical and metabolic data of 3 patients and our patient
Our patient presented with severe HTG confirmed by TG >885 mg/dL and 10 mmol/L [12]. Conservative treatment using bowel rest and hydration can lower TG level by 70% in patients with extremely high TG (>4,000 mg/dL) [13]. Plasmapharesis can acutely reduce the TG level by 50%–80% and can be instrumental if the patient has lactic acidosis, acute respiratory distress, and/or shock. This rationale was considered for this patient and 3 rounds of plasmapheresis were applied due to her metabolic/clinical decompensation. Insulin activates lipoprotein lipase, which facilitates TG degradation and should be used as a continuous infusion because it is easier to titrate along with dextrose to maintain euglycemia. Insulin can reduce TG level by 40%. In conjunction with fasting, this approach can effectively lower values by 80% in the first 24 hours of treatment [14]. Heparin, which releases endothelial lipoprotein lipase, has been used without clear benefit in HTG. Heparin has been reported to have short term effects and can increase the risk of bleeding in cases of acute pancreatitis [15]. Our patient's TG level was well controlled as she maintained a strict diet. Fenofibrate appeared to have an additive effect as it acts through the PPARα receptors and decreases the hepatic production of very low density lipoprotein. Additionally, at 4 g per day in adults, omega 3-fatty acids have an adjunctive TG lowering effect in HTG.
Our patient experienced an earlier onset of diabetes than typically seen in FPLD3. Research has suggested that development of diabetes in patients with FPLD is linked to excess subcutaneous fat accumulation in the head and neck region, beginning at puberty [16], and that individuals that harbor pathogenic variants of PPARG with reduced function in adipocyte differentiation have an estimated sevenfold increased risk of T2DM. When Hegele et al. [12] compared the phenotypes of pathogenic PPARG (FPLD3, n=8) and LMNA (FPLD2, n=51) variants, they found less adipose tissue loss, greater insulin resistance, and earlier development of diabetes (36 years vs. 41 years) in PPARG subjects. These subjects had worse hypertension and earlier presentation of polycystic ovary syndrome (PCOS), hepatic steatosis, and lower adipocytokines (leptin and adiponectin). An additional mechanism might account for the worse dysmetabolic phenotype in PPARG subjects that cannot be explained by the loss of adipose tissue. Thiazolidinediones (TZDs), a class of synthetic PPARγ agonists, promote adipogenesis and improve insulin sensitivity, which supports their therapeutic use as insulin sensitizers in patients with diabetes in FPLD3. Missense variants in PPARG are estimated to occur in 1/500 of cases, and the effects of synthetic ligands such as TZDs and tyrosine agonists on PPARγ receptors depend on the specific variant [17-19]. Agostini suggested a pharmacogenetic approach to treatment with TZDs and reported that some mild loss-of-function variants could be rescued in vitro by PPARγ agonists [20]. In contrast, Savage et al. [17] reported that severe loss-of-function PPARG pathogenic variants were not associated with clear therapeutic benefits for TZDs. Prior to initiation of piogliotazone, our patient was lost to follow up.
The c.452A>G p.Tyr151Cys pathogenic variant has been reported to cause FPLD [6,7]. This tyrosine-to-cysteine substitution is located in the region of the PPARG gene that encodes the DNA binding domain. This variant reduced DNA binding and reduced transcriptional activity, although direct dominant negative activity was absent. The second variant (c.1003G>C p.Val335Leu), initially reported by the analytical laboratory as a variant of uncertain significance, was classified by Majithia et al. as benign [8]. Visser et al. [6] described the first case of FPLD caused by the Y151C variant using the definition of severe insulin resistance (insulin requirement≥100 units/day and BMI≤27 kg/m2) in 2,500 nonobese adults with T2DM. The case was a 61-year-old European woman with a history of T2DM with onset at 49 years of age, with severe insulin resistance that required insulin at dose of 142 units/day, severe HTG with pancreatitis, hypertension, cardiovascular disease, and excess subcutaneous fat on the face, neck, trunk, and abdomen with lack of subcutaneous fat on the extremities. Akinci et al. [7] reported the p.Tyr151Cys variant in Turkish kindred (2 families). The first case was a 24-year-old female diagnosed with HTG at 15 years and diabetes at 22 years, during which she experienced metabolic complications including proteinuria, hepatic steatosis, and PCOS. There were 4 affected members in the second kindred. The proband was a 28-year-old female with new onset diabetes, HTG, and fat loss noted at 15 years of age. The mother was insulin dependent and the maternal aunt and brother had HTG. A case series of PPARγ mutations described by Akinci et al. (n=7, 77% female predominance) indicated that the average age was 28 years (range, 25–45 years); BMI 24 kg/m2 (22–25 kg/m2), average blood glucose 113 (92.5–145), and HbA1c 6% (5.5%–7.4%). All subjects had diabetes and/or prediabetes, average TG level was 228 mg/dL (range, 178–1,122 mg/dL), and leptin was 5.5 mg/dL (3–8 mg/dL). Hepatic steatosis was noted in 89% of cases, nephropathy was observed in 56%, PCOS was noted in all female patients, and fat loss was limited to the extremities. On presentation, our patient had severe HTG higher than those described by Akinci et al. [7]. She showed excessive fat on her face and neck, which was not noted in the subjects described by Akinic et al. [7] and could explain her severe insulin resistance and rapid progression to diabetes.
In conclusion, PPARG pathogenic variants should be considered in nonobese children and adolescents presenting with severe HTG and insulin resistance. Clinical acumen can help identify subtle fat loss to trigger additional testing to determine the cause of FPLD, including PPARG. Treatment with PPARy agonist TZDs should be considered because this drug class promotes adipogenesis and improves insulin sensitivity in these patients. In addition to following the metabolic and clinical parameters, delineating the changes in fat distribution with whole body MRI pre- and post-TZD can provide meaningful evidence of therapeutic efficacy.
Notes
Ethical statement
The study was provided by the Institutional Review board at New York University School of Medicine. The study met all the ethical standards as required in the study of human subjects.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Acknowledgements
The authors would like to thank Dr. Robert Hegele, Director, Blackburn Cardiovascular Genetics Laboratory, at the Robarts Research Institute for performing the genetic testing and Dr. Ambika Ashraf for her guidance in clinical management of the patient.