Steroidogenic electron-transfer factors and their diseases
Article information

Abstract
Most steroidogenesis disorders are caused by mutations in genes encoding the steroidogenic enzymes, but work in the past 20 years has identified related disorders caused by mutations in the genes encoding the cofactors that transport electrons from NADPH to P450 enzymes. Most P450s are microsomal and require electron donation by P450 oxidoreductase (POR); by contrast, mitochondrial P450s require electron donation via ferredoxin reductase (FdxR) and ferredoxin (Fdx). POR deficiency is the most common and best-described of these new forms of congenital adrenal hyperplasia. Severe POR deficiency is characterized by the Antley-Bixler skeletal malformation syndrome and genital ambiguity in both sexes, and hence is easily recognized, but mild forms may present only with infertility and subtle disorders of steroidogenesis. The common POR polymorphism A503V reduces catalysis by P450c17 (17-hydroxylase/17,20-lyase) and the principal drugmetabolizing P450 enzymes. The 17,20-lyase activity of P450c17 requires the allosteric action of cytochrome b5, which promotes interaction of P450c17 with POR, with consequent electron transfer. Rare b5 mutations are one of several causes of 17,20-lyase deficiency. In addition to their roles with steroidogenic mitochondrial P450s, Fdx and FdxR participate in the synthesis of iron-sulfur clusters used by many enzymes. Disruptions in the assembly of Fe-S clusters is associated with Friedreich ataxia and Parkinson disease. Recent work has identified mutations in FdxR in patients with neuropathic hearing loss and visual impairment, somewhat resembling the global neurologic disorders seen with mitochondrial diseases. Impaired steroidogenesis is to be expected in such individuals, but this has not yet been studied.
Highlights
Microsomal P450s need P450 oxidoreductase; its mutations cause a form of CAH. Mitochondrial P450s need ferredoxin and ferredoxin reductase (FdxR), also needed for synthesis of iron-sulfur centers. FdxR mutations cause neuropathy, but steroidogenesis in these patients has not been studied.
Introduction
Steroidogenesis is the process by which cholesterol is converted into biologically active steroid hormones, (mineralocorticoids, glucocorticoids, estrogens, progestins), and also includes the process by which a cholesterol precursor, 7-dehydrocholesterol, is converted into the biologically active form of vitamin D. Most aspects of human steroidogenesis were reviewed in 2011 [1], and some specialized aspects of steroidogenesis have been reviewed in these pages [2-4] and elsewhere [5,6]; those reviews focus on the biochemistry of the steroidogenic enzymes and the genetics and clinical manifestations of their disorders. Somewhat less attention has been directed toward the electron transfer cofactors required by steroidogenic P450 enzymes, and to the emerging understanding of their roles and the clinical diseases that arise from their mutation. The best-known and most widely seen example of such a cofactor disease is P450 oxidoreductase (POR) deficiency; much rarer disorders derive from mutations in cytochrome b5 (b5), ferredoxin (Fdx), and ferredoxin reductase (FdxR). To understand these 'cofactor diseases,' we first briefly review the biochemistry and cell biology of steroidogenesis.
Steroidogenesis
Steroidogenesis is initiated in mitochondria, where cholesterol is converted to pregnenolone by the cholesterol side-chain cleavage enzyme, P450scc, encoded by the CYP11A1 gene. Expression of CYP11A1 renders a cell 'steroidogenic,' [7] and the level of CYP11A1 transcription determines a cell's maximal steroidogenic capacity [8,9]. In adrenal and gonadal cells that produce large amounts of steroids, most cholesterol used for steroidogenesis enters the mitochondria from cytoplasmic stores with the assistance of the steroidogenic acute regulatory protein (StAR), which acts on the outer mitochondrial membrane (OMM) [9,10]. Other steroidogenic cells, notably those in the placenta, brain and skin, use cholesterol that enters the mitochondria by 'StAR-independent steroidogenesis'; this process is poorly understood, but may entail mitochondrial entry of cholesterol esters, which are freely diffusible across the mitochondrial membranes [10,11], or it may entail other proteins that substitute for StAR, such as placental MLN64 [12]. Pregnenolone may then exit the mitochondria by diffusion, or may be converted to progesterone by 3β-hydroxysteroid dehydrogenase, type 2 (3βHSD2, encoded by the HSD3B2 gene); 3βHSD2 is found in both the mitochondria and endoplasmic reticulum [13], Progesterone is then converted to androgens, estrogens, mineralocorticoids, and glucocorticoids by a series of downstream enzymes including 17α-hydroxylase/17,20-lyase (P450c17, encoded by the CYP17A1 gene), 21-hydroxylase (P450c21, encoded by the CYP21A2 gene), 11β-hydroxylase (P450c11β, encoded by the CYP11B1 gene), aldosterone synthase (P450c11AS, encoded by the CYP11B2 gene) and aromatase (P450aro, encoded by the CYP19A1 gene). The expression patterns of these enzymes differ in the various types of steroidogenic cells, accounting for their individual patterns of steroidogenesis; the genetics, cell biology, physiology, and human diseases of these enzymes have been reviewed in detail elsewhere [1].
Cytochrome P450
Cytochrome P450 enzymes are so named because they absorb light at 450 nm when the heme iron is reduced; the biochemistry of these enzymes has been reviewed elsewhere [14,15]. There are 2 types of human P450 enzymes, type 1 in the mitochondria and type 2 in the endoplasmic reticulum; the human genome encodes 57 cytochrome P450 enzymes, of which 7 are type 1 and 50 are type 2 [16]. Five enzymes involved in steroidogenesis are type 1 P450s: P450scc, P450c11β, P450c11AS, vitamin D 1α-hydroxylase (P450c1α, CYP27B1), and vitamin D 24-hydroxylase (CYP24A1). There are also 4 type 2 P450 enzymes involved in steroidogenesis: P450c17, P450c21, P450aro, and the principal vitamin D 25-hydroxylase (CYP2R1). The biochemistry, cell biology, and clinical physiology of all these enzymes have been reviewed [1,5,6]. Both types of P450 require electrons donated by reduced adenine dinucleotide phosphate (NADPH) via an electron-transport chain; the electrons must reach the heme iron atom in the P450, which mediates catalysis, but the electron-transport chains differ for the 2 types of P450 [17].
Electron transfer to mitochondrial P450s
In mitochondria, a pair of electrons from NADPH is accepted by a 54-kDa flavoprotein termed 'ferredoxin reductase' (also termed 'adrenodoxin reductase') encoded by the FDXR gene on chromosome 17q24 [18,19]. FdxR is loosely associated with the inner mitochondrial membrane. The flavin adenine dinucleotide (FAD) moiety of FdxR donates the electrons to a 14-kDa protein termed 'ferredoxin' (Fdx1, also termed 'adrenodoxin'). The same surface of the Fdx1 molecule interacts sequentially with FdxR and with the recipient mitochondrial P450 [20]: Fdx1 forms a 1:1 complex with FdxR, dissociates, then reforms an analogous 1:1 complex with the P450, thus functioning as a diffusible electron shuttle mechanism (Fig. 1). The primary RNA transcript from the FDXR gene is alternatively spliced into 2 mRNA species that can encode proteins differing by 6 amino acids [18,19], but only the shorter protein is active in steroidogenesis [21]; it is not known whether the longer form exerts an activity.
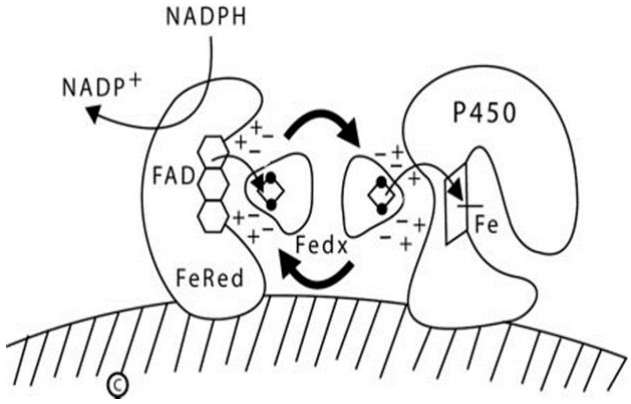
Diagram of type 1 (mitochondrial) P450 enzymes. The inner mitochondrial membrane is indicated by the hatched area; both ferredoxin reductase (FeRed) and the P450 are membrane bound, but ferredoxin (Fedx) is not. NADPH donates a pair of electrons to the flavin adenine dinucleotide (FAD) moiety of ferredoxin reductase; which then donates them to the 2Fe2S center of ferredoxin (depicted by the ball-and-stick image). The same surface of the ferredoxin molecule interacts with both the FAD of ferredoxin reductase and the redox partner binding site of the P450 by electrostatic (charge-charge) interactions. Ferredoxin thus acts as an indiscriminate electron-shuttling protein that can support the catalysis of any available type 1 P450. The electrons reach the heme iron of the P450 permitting catalysis. NADP+, nicotinamide adenine dinucleotide phosphate NADPH, reduced adenine dinucleotide phosphate.
1. Ferredoxins
Ferredoxin can carry electrons bound to its 'iron-sulfur cluster,' which contains 2 iron atoms and 2 sulfur atoms (2Fe-2S). Iron sulfur clusters can be 2Fe-2S, 3Fe-3S, or 4Fe-4S, and are found in many different proteins, such as those in the mitochondrial respiratory chain complexes I, II, and III, where they participate in electron transfer [22]. The cellular machinery that produces Fe-S clusters, which is highly conserved among eukaryotes, involves 2 major steps: assembly of the Fe-S cluster on a scaffold protein and transfer of the cluster to a recipient protein [23-26]. Defects in this process cause numerous diseases [27,28], primarily neurologic disorders such as Friedreich ataxia and Parkinson disease [29]. Ferredoxin has been known since the 1960s when it was first identified in bacteria [30]. In 1986, the laboratory of Afanasii A. Akhrem in Moscow showed that the amino acid sequences of bovine adrenal 'adrenodoxin' and liver 'hepatoredoxin' were identical [31], suggesting there was only one mammalian ferredoxin. Bovine adrenodoxin cDNA was cloned in 1985 [32]; human adrenal adrenodoxin [33] and placental ferredoxin [34] cDNAs were cloned in 1988, revealing identical sequences, showing that the same gene was expressed in both tissues; thus, finding a single gene for adrenodoxin [35] was the expected result. The single FDX1 gene is located on chromosome 11q22, with nonexpressed pseudogenes on 20q11-q12 [36,37], and is predominantly, but not exclusively, expressed in steroidogenic tissues. The human genome also contains the related FDX2 gene (formerly termed FDX1L), which encodes Fdx2. The amino acid sequences of Fdx1 and Fdx2 share only 43% identity and 69% similarity, yet have very similar 3-dimentional structures [38]; the gene sequences are sufficiently different that the FDX2 gene was not detected in studies of the chromosomal location of the FDX1 gene [36,37]. Both forms of Fdx can participate in the synthesis of Fe-S clusters [39-41], but Fdx2 is probably the more important form, especially in the central nervous system, where very little Fdx1 is found and FDX2 (on chromosome 19p13.2) is well-expressed. Because Fdx1 is abundantly expressed in steroidogenic tissues (and Fdx2 is not), Fdx1 is the form of ferredoxin that is principally involved in steroidogenesis.
A human mutation in FDX1 has not (yet) been described, but experimental deletion of the related zebrafish fdx1b gene led to defective synthesis of cortisol and androgens [42,43]. However, there are important differences in the steroidogenesis of zebrafish and human beings, hence the zebrafish results may not indicate what the effects of a human FDX1 mutation would be. However, mutations in both FDX2 and FDXR have been reported recently; mutations in both of these genes yielded neurologic impairments, apparently related to impairment of the synthesis of Fe-S clusters, and yielding global mitochondrial dysfunction. Two studies reported a novel mitochondrial muscle myopathy without optic atrophy or reversible leukoencephalopathy (MEOAL) in patients with FDX2 mutations. In the initial report, a 15-year-old girl with MEOAL was born to consanguineous parents; whole exome sequencing identified a homozygous missense mutation in the initiation codon of the FDX1L (FDX2) gene [44]. The Fdx2 protein was essentially undetectable in a muscle biopsy and in cultured fibroblasts. Six similar patients from 2 families were homozygous for a Fdx2 missense mutation [45]. RNA and protein blotting studies suggested the mutant protein was unstable. The MEOAL phenotype is consistent with impaired formation of Fe-S clusters, which are required by several mitochondrial respiratory chain proteins.
2. Ferredoxin reductase
In addition to Fdx1 and Fdx2 (and many other proteins), the synthesis of Fe-S clusters requires FdxR [23-26]. This recently discovered role of FdxR in Fe-S cluster biosynthesis explains the formerly confusing observation that low levels of mRNA encoding FdxR were found in all tissues, although expression in steroidogenic tissues was about 100-fold greater [46]. The human genome contains only one copy of the FDXR gene encoding FdxR. Because both ferredoxins play a role in the biogenesis of Fe-S centers and there is only one FDXR gene, one would presume that FDXR mutations would also affect Fe-S synthesis and result in a similar phenotype. Knockdown of FDX1, FDX2, or FDXR in human cell lines diminished the synthesis of Fe-S clusters and impaired the activity of several enzymes that rely on Fe-S clusters for activity, as well as depleting cytosolic iron and causing mitochondrial iron overload [40,41]. Thus, interference with FDX1, FDX2, or FDXR can disrupt the synthesis or assembly of Fe-S clusters and disrupt cellular iron homeostasis.
Consistent with these observations, 45 patients in 33 families have now been described with mutations in FDXR [47-52]. Most of the families are reportedly unrelated to one another, but the high frequency of specific mutations in some studies suggests ether unknown consanguinity or local founder effects. Thus, among the 13 families (26 alleles) in one study, 11 alleles carried the missense mutation R392W [48], and among 8 families (16 alleles) in another study, 5 alleles carried the missense mutation P372H [52], yet neither of these mutations was detected in any other study. It is noteworthy that all reported patients have at least one FDXR allele that has a missense mutation that retains partial activity; no patient has been reported with null FDXR alleles on both parental chromosomes. The same situation is seen in patients with POR deficiency (see below). Such observations suggest that homozygosity (or compound heterozygosity) for null alleles may be lethal in embryonic or fetal life.
The most consistent clinical findings in FdxR deficiency are optic atrophy, neuropathic hearing loss, and developmental delay; some patients had mild movement disorders and even Leigh syndrome with infantile-onset encephalopathy and death. Mice possessing a naturally occurring homozygous R389Q mutation in Fdxr [53] (corresponding to human R392Q) have a progressive gait disorder, decreased visual acuity, and levels of Fdxr activity at 33%–50% of normal, depending on the tissue assessed [48]. Consistent with these genetic observations, primary cultures of patient fibroblasts had reduced FdxR activity and increased production of reactive oxygen species [48], and siRNA-mediated knockdown of FdxR in HeLa cells and in human K562 erythroid cells led to iron overload [40]. Thus, clinical, genetic, biochemical and cell biologic data show that deficiency of FdxR results in a mitochondrial disorder, primarily manifested in the central nervous system, that shares many features with Fdx2 deficiency and other mitochondrial disorders. However, from an endocrine perspective, the most remarkable feature of all these reports is the absence of any studies directed toward the obligatory role of FdxR in steroid hormone (and vitamin D) synthesis. Future studies should include clinical investigation into adrenal reserve (e.g., by performing adrenocorticotrophic hormone [ACTH] tests) and cell biologic studies of steroidogenesis (e.g., by transfecting nonsteroidogenic cells with vectors expressing a mitochondrial P450 plus either wild type (WT) or mutant FdxR). One might speculate that patients with FDXR mutations that retain partial activity will have compensated adrenal insufficiency, as seen in the nonclassic forms of lipoid congenital adrenal hyperplasia (CAH) [54] and P450scc deficiency [55].
Electron transfer to microsomal P450s
In the endoplasmic reticulum ('microsomes') NADPH donates electrons to the 82-kDa, 2-flavin protein POR, which then donates them to the microsomal steroidogenic P450 enzymes (P450c17, P450c21, P450aro, and CYP2R1) and to the other 46 microsomal P450 enzymes that are involved in the synthesis of eicosanoids and leukotrienes, and in the metabolism of drugs and xenobiotics [16]. POR also donates electrons to squalene monoxygenases, fatty acid elongase, heme oxygenase, and b5 [56]. This is a 2-step process. POR has 2 distinct domains that resemble the wings of a butterfly: one wing contains a FAD moiety, and the other contains a flavin mononucleotide (FMN) moiety. These domains are joined by a 'hinge' domain [57]. Before interacting with NADPH, POR is in a relaxed, open state. When electrons from NADPH bind to the FAD moiety, POR undergoes a conformational change bringing the 2 'wings' close together, permitting the electrons to travel from the FAD to the FMN; when this happens, the POR then 'relaxes,' permitting the FMN domain to interact with the redox-partner binding site of the P450, thus transferring the electrons to the heme iron atom in the P450, which mediates catalysis (Fig. 2) [58].
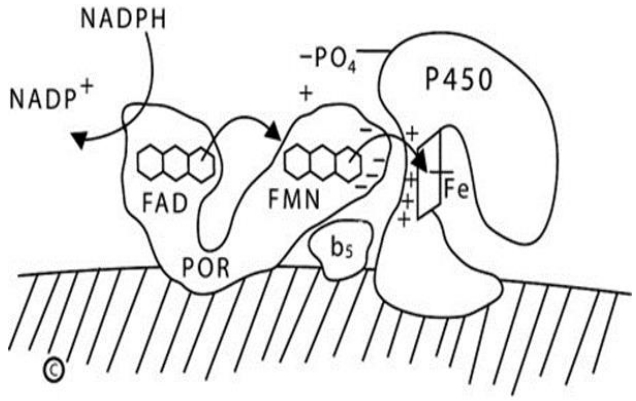
Diagram of type 2 (microsomal) P450 enzymes. The endoplasmic reticulum membrane is indicated by the hatched area; both P450 oxidoreductase (POR) and the P450 are membrane-bound. NADPH interacts with the flavin adenine dinucleotide (FAD) domain of POR and donates a pair of electrons to the FAD moiety. Electron receipt elicits a conformational change, permitting the isoalloxazine rings of the FAD and flavin mononucleotide (FMN) moieties to come close together, permitting the electrons to transfer from the FAD to the FMN. Electron receipt by the FMN reverts the POR protein to its original, open conformation, permitting the FMN domain to interact with the redox partner binding site of the P450 by electrostatic charge interactions. The electrons reach the iron atom of the heme group of the P450, permitting catalysis. For some reactions catalyzed by some P450 enzymes, notably the 17,20-lyase activity of human P450c17, cytochrome b5 acts allosterically to promote increased activity. NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced adenine dinucleotide phosphate.
1. POR deficiency
The participation of POR in so many essential biochemical functions would suggest that POR deficiency might be lethal, and POR-knockout mice die during fetal development [59,60]. Nevertheless, POR deficiency is compatible with human life, causing a severe skeletal malformation syndrome and a form of CAH characterized by partial (and variable) deficiencies in the activities of P450c17, P450c21, and P450aro [61,62]. The associated skeletal malformation syndrome is a form of Antley-Bixler Syndrome (ABS), which is characterized by craniosynostosis, brachycephaly, radio-ulnar or radio-humeral synostosis, bowed femora, arachnodactyly, midface hypoplasia, proptosis, and choanal stenosis. The ABS phenotype can be caused either by recessive POR mutations or by dominant, gain-of-function mutations in fibroblast growth factor receptor 2 (FGFR2) [62]. When ABS is seen in association with abnormal steroids and ambiguous genitalia in either sex, the cause is POR; when ABS is seen without disordered steroidogenesis or genital development, the cause is FGFR2. The ABS phenotypes resulting from POR or FGFR2 mutations are indistinguishable, even though the mutations in the POR and FGFR2 genes segregate completely [62]. The ABS skeletal phenotype in patients with POR deficiency probably results from diminished activity of CYP26B1, the POR-dependent microsomal enzyme that degrades retinoic acid [63]. Studies of 2 families with CYP26B1 mutations and the recreation of their mutations in transgenic mice and zebrafish show that retinoic acid must be degraded locally in the embryonic connective tissues that form skeletal joints and sutures; POR deficiency disrupts CYP26B1 activity, accounting for the skeletal phenotype. Other mechanisms, including defective signaling by hedgehog proteins secondary to a POR-associated defect in cholesterol synthesis may also play a role. Dozens of human POR mutations have now been described, affecting various P450 enzymes to differing degrees, explaining the great variability in the clinical and hormonal findings in POR deficiency.
POR deficiency has different steroidal phenotypes, depending on the specific POR mutation(s). Impaired P450c21 activity may lead to circulating concentrations of 17OH-progesterone that are high enough to trigger newborn screening programs for CAH [61,62,64,65]. Most patients with POR deficiency have normal electrolytes and normal basal cortisol concentrations that respond poorly to ACTH, indicating compensated adrenal insufficiency [61,62,66,67]. POR deficiency, usually when sufficiently severe to result in ABS, can cause genital ambiguity: males may be underdeveloped and females may be virilized. Androgen synthesis is typically impaired in POR deficiency by the effect of POR mutations to diminish the 17,20-lyase activity of P450c17, as this activity is especially sensitive to impaired electron transport [17,68,69]. Thus, males with POR deficiency are hypoandrogenic; those with severe POR defects have underdeveloped external genitalia, and those with milder defects may only have infertility [62,64,65]. Females with POR deficiency may be partially virilized by 2 mechanisms. First, some (but not all) POR mutations that cause ABS will result in partial deficiency of placental aromatase (P450aro) activity, resulting in fetal virilization similar to that seen with P450aro deficiency [61,62]. This is the usual outcome among infants carrying the R457H mutation (prevalent in Japan), but not with the A287P mutation (prevalent in Europe) [61,62,66,70]. Women carrying a fetus with POR R457H have low estriol levels [71,72], and the R457H mutant does not support P450aro activity in vitro, whereas the A287P mutant retains full in vitro activity with P450aro [73]. Second, POR deficiency drives steroid precursors into the alternative "backdoor" pathway of androgen biosynthesis [74], converting fetal 17OHP to androgens [72,75]. The relative importance of these 2 mechanisms for virilizing the fetus with POR deficiency varies with the specific POR mutation involved.
2. POR genetics and regulation
The single human POR gene, located on chromosome 7, consists of 15 protein-coding exons and a first untranslated exon that lies ~39 kb upstream and initiates transcription. The POR gene is highly polymorphic: among 842 persons from the San Francisco area who identified themselves as African-American (AA), Caucasian-American (CA), Mexican-American (MA), or Han Chinese (Asian) American (AS), there was a high degree of polymorphism, including 140 single nucleotide polymorphisms present in >1% of 1 of the 4 populations [76]. By far the most common polymorphism resulted in an amino acid change (A503V), which was found on 19.1% of AA alleles, 26.4% of CA alleles, 31.0% of MA alleles and 36.7% of AS alleles (overall incidence of 27.9% of all alleles) [76]. There did not seem to be any selection for this variant, as it was in Hardy-Weinberg equilibrium.
Rat POR transcription requires thyroxine acting through a thyroid-response region at bases -564 to -536 [77,78]. Computa - tional searches of 10 kb of 5' flanking DNA in the human POR gene identified no conserved regions >2.5-kb upstream, and no apparent transcription factor binding sites more than 0.9-kb upstream from the transcriptional start site [79]. Most basal transcriptional activity in the human POR promoter lies within 325 bp from the untranslated exon. This region contains common polymorphisms at -208, -173, and -152 [76]. Among these 3 polymorphisms, only the one at -152 reduced transcription (by 65% in human adrenal NCI-H295A cells) [79]. Electrophoretic mobility shift assays identified binding of Smad3/Smad4 between -249 and -261 and binding of thyroid hormone receptor-β (TRβ) at -240/-245, but did not detect proteins binding to either the WT or polymorphic sequence at -152. Chromatin immunoprecipitation confirmed that Smad3, Smad4, TRα, TRβ, and estrogen receptor-α (ERα) bound the POR promoter between -374 and -149. Co-expression of these transcription factors and POR promoter-reporter constructs followed by treatment with estradiol or triiodothyronine showed that triiodothyronine exerts major tropic effects via TRβ, and that TRα, ERα, Smad3 and Smad4 exert lesser, modulatory effects [79]. Thus, TRβ and Smad3/4 are key factors in human POR expression, and the common polymorphism at -152 may play a role in genetic variation in steroid biosynthesis and drug metabolism.
3. POR and drug metabolism
Because POR is required by all drug-metabolizing P450 enzymes, there has been considerable interest in the potential impact of POR allelic variations on drug metabolism. Among the 140 human POR SNPs, only 2 were found in >2% of the population: these were the coding sequence variant A503V and the C to A change at -152 in the promoter [76]. A503V was the most common allelic variant. The activity of POR A503V has been assayed by measuring the parameter Vmax/Km for the activity of a P450 enzyme compared to P450's activity with WT, control POR. The activity of A503V to support catalysis by steroidogenic enzymes varied: A503V had 68% of WT ability to support the 17α-hydroxylase activity of human P450c17 and 58% of WT ability to support its 17,20-lyase activity [62,76]. By contrast, A503V had 80% of WT activity to support the 21-hydroxylation of progesterone by human P450c21, and 95% of WT ability to support the 21-hydroxylation of 17OH-progesterone [80]. Thus, POR A503V may contribute to reductions in androgen synthesis by P450c17 but has minimal impact on the activity of P450c21. Furthermore, these studies show that the ability of a POR variant to support the activity of one P450 enzyme does not predict its ability to support the activity of other P450 enzymes.
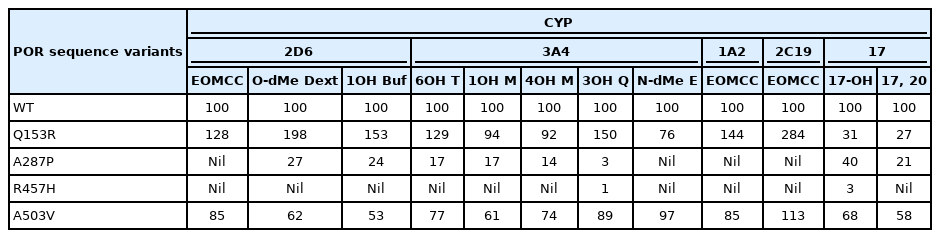
A survey of 35 POR variants with the human hepatic enzymes CYP1A2 or CYP2C19 found substantial differences among the POR variants studied, including some (notably Q153R) that increased activity [81]. The human hepatic enzymes CYP3A4 and CYP2D6 respectively metabolize ~45% and ~25% of clinically-used drugs [82,83]; studies addressed their activities with the common A503V variant, the common disease-causing mutants A287P and R457H, and the Q153R gain-of-function mutant. CYP3A4 can metabolize a huge array of drugs because it can alter its shape to accommodate the substrate. Crystallography shows that human CYP3A4 has the same fold as other P450 enzymes, but that its substratebinding pocket is highly distensible: it has a volume of ~520 Å3 in the absence of substrate or in association with metyropone (212 Da) or progesterone (318 Da) [84], but expands to ~2,000 Å3 when binding erythromycin (734 Da) [85]. To survey the potential impact of such substrate-induced conformational changes in CYP3A4 on electron donation from POR, the ability of WT, Q153R, A287P, R457H, and A503V POR to support catalysis by CYP3A4 was assessed using 4 substrates: testosterone (288 Da), midazolam (326 Da), quinidine (324 Da) and erythromycin (734 Da), representing drugs of different sizes and chemical classes (Table 1) [86]. When the 6β-hydroxylation of testosterone by CYP3A4 was supported by POR Q153R, it had 129% of WT activity. With POR A287P it had 17% of WT activity, with POR R457H activity was barely detectable, and with POR A503V it had 77% of WT activity. CYP3A4 metabolizes midazolam by both 1-hydroxylation and 4-hydroxylation. When supported by POR Q153R, CYP3A4 had 92%–94% of WT activity for these activities; POR A287P supported 14%–17% of WT activity, R457H supported minimal activity, and POR A503V supported 61%–74% activity. Quinidine has 2 fused rings, and is 3-hydroxylated by CYP3A4; when supported by POR Q153R, CYP3A4 activity was 150% of WT activity, POR A287P, and R457H supported barely detectable activity, and POR A503V supported 89% of WT activity. CYP3A4 catalyzes N-demethylation of erythromycin; to accommodate erythromycin, the substrate-binding pocket of CYP3A4 expands substantially. POR Q153R, which shows increased activity in other circumstances, had only 76% of WT activity to support CYP3A4 metabolism of erythromycin. A287P and R457H supported minimal activity, but A503V had full WT activity to support the metabolism of erythromycin by CYP3A4. Thus the ability of different POR variants to support the activity of CYP3A4 varies with the substrate: both substrate size (as evidenced by erythromycin) and chemical structure (as evidenced by midazolam) are important [86]. Similar studies examining the ability of POR variants Q153R, A287P, R457H, and A503V to support 3 activities catalyzed by CYP2D6 (activation of EOMCC, O-demethylation of dextromethorphan and 1-hydroxylation of bufuralol) again showed decreased activity of A503V [87]. Thus, A503V shows reduced activity to ~60% of WT in many, but not all assays, underscoring the need to test each reaction of interest (Table 1). These data have been reviewed previously [88].
Cytochrome b5
1. Promotion of P450 activities by b5
Cytochrome b5 (b5), encoded by the CYB5A gene on chromosome 18p13, is a small heme-containing protein found in 3 forms [89,90]. The 98 amino acid cytosolic form is mainly expressed in erythrocytes, where it reduces methemoglobin to hemoglobin, while the 134 amino acid endoplasmic reticulum form and the 146 amino acid form found on the OMM are widely expressed, including in steroidogenic tissues [91]. Cytochrome b5 has 2 domains: one binds heme and the other forms a structural core, from which the C-terminal membrane-anchoring helix extends. The surface of the heme-binding domain has numerous negatively charged residues that facilitate electrostatic interactions with other proteins. Cytochrome b5 can augment some P450 activities, possibly involving direct electron transfer from b5 to the P450 [92]. However, some of the actions of b5 can be observed with apo-b5, which lacks its heme group and hence cannot transfer electrons [93]. With human P450c17, b5 selectively stimulates 17,20-lyase activity but has negligible effects on 17-hydroxylase activity [94,95].
Substantial evidence indicates that b5 enhances the interaction of P450c17 and POR, promoting more efficient electron transfer [94,95]. This mechanism would account for b5 having no effect on the Km of P450c17, while increasing the Vmax of the 17,20-lyase reaction [69,94,95]. It is consistent with data showing that excess POR increases 17,20-lyase activity in the absence of b5 [96,97], and that mutations in the POR binding site of P450c17 selectively reduced 17,20-lyase activity [69]. Additionally, when the molar ratio of b5 to P450c17 is fixed, 17,20-lyase activity shows a dose-response to added POR [95]; a dose-response was also observed in the absence of b5, but 17,20-lyase activity was always higher in the presence of b5, confirming that b5 augments 17,20-lyase activity by facilitating electron transfer from POR [95]. Alternatively, b5 could promote 17,20-lyase activity by inducing a conformational change in P450c17 that might promote the interaction of steroid carbon number 20 (C20), rather than C17, with the heme iron of P450c17 [98]. Such b5-induced conformational changes with changes in activity have been reported with other P450 enzymes. A modest b5-induced change in the conformation of the substrate-binding pocket of P450c17 has been reported [99], and at least one mutation in the active site of P450c17 has been reported to impair 17,20-lyase activity selectively [100]. Thus, both mechanisms appear to be involved.
The 17,20-lyase activity of P450c17 can also be increased by the serine/threonine phosphorylation of P450c17 [101,102], probably catalyzed by p38α, a cAMP-dependent mitogen-activated protein kinase [103], apparently increasing the association of P450c17 with POR [95], Genetic and biochemical studies implicate positively charged residues in P450c17, including R347, R358 (and perhaps R449 and K89) in its interaction with b5 [68,69]; correspondingly, residues E48 and E49 of b5 are required for high 17,20-lyase activity [104]. Thus, the regulation of 17,20-lyase activity, and consequently of androgen production, depends on factors that facilitate the flow of electrons to P450c17: high concentrations of POR, the presence of b5, and serine phosphorylation of P450c17 [105].
2. Mutation of b5
Mutations of P450c17 R347 or R358, which disrupt electron transfer from POR, were the first form of 17,20-lyase deficiency to be demonstrated genetically and biochemically [68]. This prompted a broader search for mutations in factors affecting electron transport as potential causes of 17,20-lyase deficiency. The first case of b5 deficiency was reported in a patient with methemoglobiemia and ambiguous genitalia, but unfortunately no studies of adrenal or gonadal steroidogenesis were reported [106,107]. Methemoglobinemia is a predictable consequence of b5 deficiency, as the reduction of methemoglobin is the principal physiologic role of b5, and the common cause of methemoglobiemia is deficiency of b5 reductase. More recently, a consanguineous 46,XY infant was reported who had micropenis, bifid scrotum, scrotal hypospadias, and homozygous b5 mutation W27X [108]. By age 3 months he had hypergonadotropic hypogonadism with low adrenal and gonadal C19 steroids and a normal cortisol response to ACTH; the methemoglobin level was 4-fold above the upper limit of normal, but clinical methemoglobinemia was not apparent. Cytochrome b5 residues E48 and E49, which are required to stimulate 17,20-lyase activity, are absent with the W27X mutation; the residues required for the reduction of methemoglobin have not been mapped, but this activity should require heme binding. Since these reports, a small number of additional patients have been characterized [109-111]. Thus, b5 deficiency is an important cause of 17,20-lyase deficiency that does not appear to affect cortisol synthesis, whereas specific P450c17 mutations and the one reported POR mutation that presented as 17,20-lyase deficiency [64] may have residual cortisol synthesis [105].
Conclusion
Disorders in the factors that participate in electron transfer from NADPH to cytochrome P450 enzymes are a newly recognized group of disorders of steroidogenesis. Mutations in POR, first described in 2004, are fairly common and are now well-characterized clinically, genetically and biochemically. Mutations in b5 were first described in 2010, causing isolated 17,20-lyase deficiency, but this remains one of the rarest disorders in steroidogenesis. Mutations in FdxR were first reported in 2017 and cause neuropathic hearing loss and visual impairment, but potential (probable) steroidogenic consequences have not been reported or sought. As Fdx and FdxR are essential components in the synthesis of Fe-S clusters, a neurologic phenotype is not surprising. Careful clinical studies of adrenal and gonadal steroidogenesis in these patients are needed, as are in vitro studies of steroidogenesis with recapitulation of the known FdxR mutations in conjunction with a mitochondrial P450 enzyme such as P450scc.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
Work done in the author's laboratory was supported by grants from the National Institutes of Health and from private foundations.