Highlights
Second-generation somatostatin analogues might be effective in reducing insulin secretion in congenital hyperinsulinism (CHI). We report the first use of pasireotide in a boy with severe therapy-resistant CHI, resulting in slightly improved glycemic control without side effects.
Introduction
Congenital hyperinsulinism (CHI) is a rare disorder characterized by inappropriate insulin secretion from pancreatic beta cells in the presence of a low blood glucose concentration. CHI is the most common cause of recurrent and persistent hypoglycemia in neonates and children [1]. Clinical severity can range from mild to severe depending on the underlying cause. At least 15 genes associated with CHI have been identified: ABCC8, KCJN11, GLUD1, GCK, HADH, SLC16A1, UCP2, HNF4A, HNF1A, HK1, PGM1, PPM2, CACNA1D, FOXA2, and EIF2S3. [1] Inactivating mutations in the ABCC8 and KCJN11 genes account for the majority of cases and are associated with the most severe and therapy-resistant forms. These 2 genes encode the 2 subunits of the ATP-sensitive potassium channel in beta cells.
The main treatment goal in CHI is to prevent hypoglycemia and consequent neurological damage. Current treatment protocols are primarily based on dietary and pharmacological interventions [2,3]. Further, surgery might be necessary in specific cases of focal or severe diffuse hyperinsulinism. Initial medical treatment consists of diazoxide; based on the clinical response, CHI patients are classified as either diazoxide responsive or unresponsive. Because diazoxide inhibits insulin release by opening the ATP-sensitive potassium channels, unresponsiveness is typically seen in patients with homozygous or compound heterozygous ABCC8 or KCJN11 mutations. Second-line treatment with octreotide can be applied in diazoxide-unresponsive cases [2,3]. Other proposed (off-label) medicines in patients with severe CHI include long-acting octreotide analogues, continuous glucagon infusion, sirolimus, nifedipine, and glucagon-like peptide-1 inverse agonists [4]. In severe diffuse CHI, response to medical treatment is often limited, and near-total pancreatectomy might be needed [2,5]. However, with respect to pancreatic surgery, a restrained policy is recommended as the severity of CHI tends to decrease over time, potentially allowing a better response to treatment. Furthermore, near-total pancreatectomy can cause exocrine insufficiency and result in long-term insulin-dependent diabetes [5].
Case report
1. Initial presentation
The patient presented after birth and after an uncomplicated pregnancy of 38 weeks and 3 days as the fourth child of consanguineous Pakistani parents. There was no gestational diabetes. The boy's birthweight was 5,085 g (standard deviation score [SDS] >2.5), and vaginal delivery was complicated by shoulder dystocia. Apgar scores were 4 and 7 after 1 and 5 minutes, respectively. Directly postpartum, blood glucose levels were unmeasurably low for 3.5 hours despite administration of multiple intravenous glucose boluses and increased carbohydrate intake. Within 48 hours after birth, the carbohydrate intake had been increased to 21 mg/kg/min. A diagnosis of hyperinsulinism was biochemically confirmed by identifying an elevated insulin level of 415 pmol/L (reference range, 12–96 pmol/L) measured during hypoglycemia (glucose 1.5 mmol/L). On the second day of life, oral diazoxide treatment was started. Because no response to diazoxide (maximum dose 20 mg/kg/day) and an additional increase in carbohydrate intake (up to 26 mg/kg/min) were observed, intravenous octreotide treatment was started on the ninth day of life. As blood glucose level did not increase on continuous octreotide (maximum dose 15 μg/kg/day), continuous glucagon infusion was added on the 11th day of life, initially subcutaneous but changed to intravenous at a maximum dose of 20 μg/kg/hour after insertion of a central venous line.
2. Genetic diagnosis
At the age of 3 weeks, diagnosis of diffuse CHI was genetically confirmed. The boy was a carrier of a pathogenic homozygous ABCC8 mutation that consisted of a duplication of 2 nucleotides and was predicted to cause a frameshift and a premature stop codon at position 6 in the new reading frame (c.597_598dup; p.(Thr200Argfs*6)). Since the father was in Pakistan and unable to travel to the Netherlands, genetic testing of the parents was not performed. The medical history of the patient's parents and 4 siblings did not reveal any signs or symptoms of hyperinsulinism. The presence of larger deletions or insertions in ABCC8 was excluded using Multiplex Ligationdependent Probe Amplification analysis.
3. Treatment up to first surgery
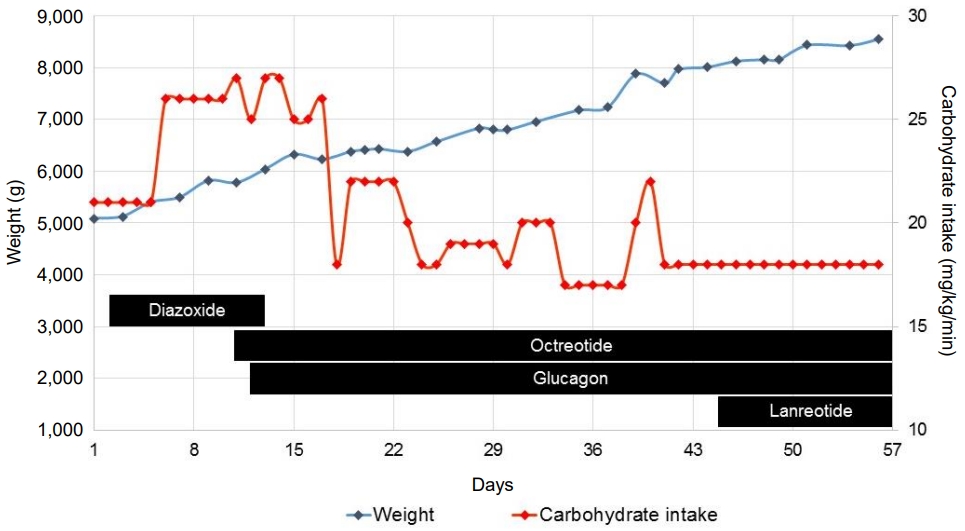
A schematic overview of pharmacological treatment (diazoxide, octreotide, glucagon, and lanreotide), carbohydrate intake, and weight progression of the boy during his first 2 months of life is shown in Fig. 1. Despite high-dose octreotide and continuous glucagon infusion, a high carbohydrate intake (18 mg/kg/min) was required to prevent hypoglycemia. This led to excessive weight gain (Fig. 1). Because lowering the continuous octreotide infusion immediately resulted in hypoglycemia, the long-acting somatostatin analogue somatuline autogel (lanreotide) was started at 6 weeks of age. Since this did not lead to improvement, a laparoscopic partial pancreatectomy was performed at the age of 8 weeks (tail resection, approximately 60% remaining pancreatic tissue).
4. Treatment up to second surgery
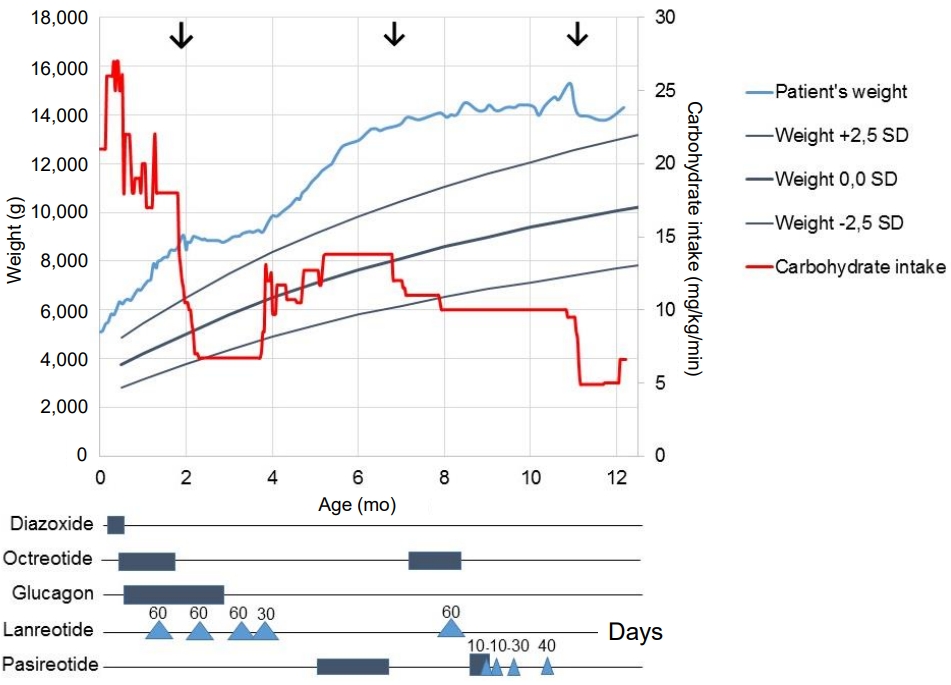
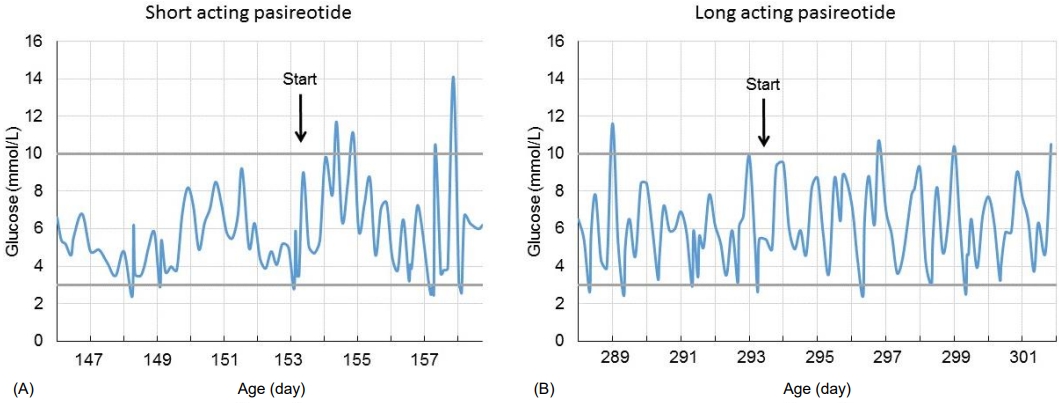
The initial postoperative response was promising, and one month after surgery, glucagon was stopped and feeding frequency was reduced to every 3 hours, with a carbohydrate intake of 7.5 mg/kg/min. Postoperatively, lanreotide treatment (60 mg subcutaneous [s.c.] every 4 weeks) was continued. Unfortunately, 2 months after surgery, an unexpected decrease in blood glucose concentration occurred. Continuous drip feeding using a gastrostomy tube was required, and carbohydrate intake was increased to 12 mg/kg/min, after which weight gain increased again (Fig. 2). In an attempt to avoid further surgery and after consulting international CHI experts, an offlabel trial with the second-generation somatostatin analogue pasireotide was started, with parental consent. Initially, short-acting s.c. pasireotide injections were given in a dose of 0.15 mg twice daily. This led to high blood glucose concentration shortly after injection (Fig. 3). Because of large fluctuations in glucose concentrations, with high values noted shortly after injection and low values just before injection, the dose and frequency of pasireotide injections were increased from twice daily at 0.15 mg to 4 times daily at 0.3 mg to avoid hypoglycemia. Although this resulted in less fluctuation, glucose levels were often at the lower limit of the desired target range (around 3.0 mmol/L). Therefore, second laparoscopic partial pancreatectomy was performed at the age of almost 7 months (body resection; 35% remaining pancreatic tissue).
5. Treatment up to third surgery
The result of this second surgical intervention was rather disappointing. Carbohydrate intake could not be reduced (carbohydrate intake of 10 mg/kg/min, feeding every 2 hours), and octreotide (40 μg/kg/day, continuous s.c.) and lanreotide (60 mg s.c. every 4 weeks) were restarted. Because pasireotide seemed to have a better effect than lanreotide, it was restarted at the age of almost 9 months (short-acting pasoreotide, 0.3 mg s.c. 3 times daily). Similar to the first period of short-acting pasireotide injections, glucose levels fluctuated, and short-acting pasireotide was substituted with long-acting pasireotide (starting dose 10 mg every 28 days). Within a few weeks, the dose was increased to 40 mg per 28 days (adult dose for acromegaly), resulting in fewer hyperglycemic episodes (Fig. 3). Potential side effects of pasireotide (elevated liver enzymes, hypothyroidism, and adrenal insufficiency) were monitored and did not occur. Although carbohydrate intake remained at 10 mg/kg/min during pasireotide treatment, the boy still had periods of hypoglycemia. Minimally invasive laparoscopic near-total pancreatectomy was considered as the only remaining treatment option and was performed at the age of almost 11 months (approximately 5% remaining tissue). Pathologic evaluation after (each) pancreatectomy showed diffuse hyperplasia of the pancreatic beta cells and insulin-positive cells with immunohistochemistry, which fits with the diagnosis of severe diffuse hyperinsulinism.
6. Clinical course after third surgery
The long-acting pasireotide treatment was stopped postoperatively. To test for potential pasireotide-induced central adrenal insufficiency, low dose (1 μg) and classic (36 μg/kg) adrenocorticotropic hormone (ACTH) tests were performed, and both results were normal. The patient's carbohydrate intake could be reduced to 6.5 mg/kg/min, the interval between feedings could be increased to 4 hours, and weight gain started to normalize. Just after his first birthday, the boy was discharged from the hospital.
At the last outpatient clinic visit, at the age of 1.5 years, the boy still occasionally experienced glucose levels at the lower limit of the desired target range (around 3.0 mmol/L), required oral feeding every 3 hours during the day (enriched with maltodextrin), and feedings every 4 hours during the night via a gastrostomy tube to prevent hypoglycemia. His current carbohydrate intake was 6.5 mg/kg/min. No progressive weight gain had occurred since the last surgery, and his weight SDS improved further (weight SDS, +2.79; height SDS, +0.42). While he had shown mildly impaired psychomotor development during hospitalization, at 1.5 years of age, his psychomotor development appeared normal. Magnetic resonance imaging of the brain, performed at one month of age, showed no signs of hypoglycemic damage, and has not been repeated. Since discharge, one symptomatic hypoglycemic episode occurred, resulting in a seizure. The mild impairment in psychomotor development during hospitalization was probably of multifactorial origin: hypoglycemia, severe obesity with impaired physical activity, and under-stimulation during longterm hospitalization.
Discussion
Here we report the first use of pasireotide in the treatment of a child with CHI. In this case, a homozygous ABCC8 mutation led to a severe, therapy-resistant form of CHI. Although pasireotide somewhat improved glycemic control, it was not sufficient to avoid near-total pancreatectomy.
Inactivating homozygous or compound heterozygous ABCC8 and KCJN11 mutations are associated with severe diffuse CHI that is usually unresponsive to pharmacological treatment and requires subtotal pancreatectomy. Since diazoxide inhibits insulin secretion by opening the ATP-sensitive potassium channels, it is not surprising that these mutations result in diazoxide unresponsiveness.
Second-line treatment consists of somatostatin analogues. Somatostatin is one of the main inhibitors of endocrine and exocrine hormone secretion. Somatostatin exerts its effect by binding to its receptor, of which there are 5 subtypes (SSTR1-5). Inhibition of insulin secretion is mainly mediated by SSTR2 and SSTR5. Several somatostatin analogues have been developed to treat conditions characterized by overproduction of hormones, such as acromegaly and Cushing disease. Current CHI treatment protocols include 2 long-acting somatostatin analogues, octreotide long-acting release (sandostatin-LAR) and somatuline autogel (lanreotide) [6,7]. In a multicenter study including 27 patients, these long-acting somatostatin analogues were found to beffective in achieving normoglycemia. The most common side effect was liver enzyme elevation, observed in 37% of cases [8]. Sandostatin-LAR and lanreotide belong to the first-generation somatostatin analogues and mainly bind to SSTR2 and SSTR5. Pasireotide is a second-generation somatostatin analogue that targets 4 of 5 somatostatin receptors, with the highest affinity for SSTR1 and SSTR5 [9]. Pasireotide was first introduced for treating Cushing disease and is known to cause severe hyperglycemia as a side effect due to inhibition of insulin secretion. Pasireotide is an approved treatment for Cushing disease and acromegaly; based on its affinity for SSTR5, it has been proposed as a promising new treatment for CHI [5]. Pasireotide has a much higher affinity for SSTR5 than the first-generation somatostatin analogues [9] and might be more effective in controlling hyperinsulinism. In addition, pasireotide causes less inhibition of glucagon secretion through SSTR2 than first-generation somatostatin analogues [10]. In a previous case report of an adult woman with nesiodioblastosis, pasireotide was successfully used to control persistent hypoglycemia without reducing ACTH and cortisol levels [11].
In our patient with severe, therapy-resistant CHI, pasireotide was used in a last attempt to avoid near-total pancreatectomy. Although the treatment period was rather short (2 months), the boy did seem to show a response as short-acting pasireotide injections lead to large fluctuations in blood glucose, including hyper- and hypoglycemia (Fig. 3). The use of long-acting pasireotide (maximum dose used was 40 mg every 28 days) (Fig. 3) reduced the hyperglycemic episodes. Although we did not notice adverse effects, hypocortisolism and liver dysfunction are possible after prolonged use. Unfortunately, the effect of pasireotide in our patient with severe CHI was insufficient to avoid definitive pancreatic surgery. In 2014, the same homozygous ABCC8 duplication was described in another severe CHI case as a novel mutation [12]. Unfortunately, in that case, frequent hypoglycemic episodes led to developmental delay [12]. We were able to minimize hypoglycemia in our patient, and at present, the boy shows no signs of neurological damage. However, even after surgery, he requires a higher-than-normal carbohydrate intake and has a fasting tolerance of only 4 hours. After near-total pancreatectomy, during the middle term, up to 60% of patients have hypoglycemic episodes requiring pharmacological treatment and frequent feedings [13].
Our patient was hospitalized during his entire first year of life. Since even severe forms of CHI have been reported to become milder over time, possibly due to apoptosis of hyperinsulinemic beta cells, the current opinion is to try to avoid surgery. That is why initially only partial rather than near-total pancreatic resection was performed. However, in hindsight, in such a severe, diffuse form of CHI, near-total pancreatectomy might have been a better choice to prevent long-lasting hospitalization.
In summary, in this case of severe CHI, a short period of pasireotide treatment slightly improved glycemic control without side effects. Unfortunately, the effect of pasireotide on glycemic control was insufficient to prevent near-total pancreatectomy. Potential side effects during prolonged treatment, such as decrease in ACTH and cortisol levels, require further investigation.