Highlights
We report 2 patients with CHI caused by novel DNA variants in ABCC8. Intestinal malrotation occurred after diazoxide treatment in one patient. Genetic analysis should be performed in all patients and family members that present with CHI to define the best treatment approaches.
Introduction
Congenital hyperinsulinism (CHI) is a rare disease that can result in potentially life-threatening hyperinsulinemic hypoglycemia (HH), caused by inadequate insulin secretion by pancreatic ÎČ-cells. The incidence is estimated to be 1:50,000 live births, and 1:2500 live births in communities with founder mutations [1]. Rapid diagnosis and treatment are essential to prevent persistent or recurrent hypoglycemia and neurological damage [2-5]. CHI can be classified as either transient or permanent and has 3 histologic subtypes: focal, diffuse or atypical CHI. Standard treatment for focal CHI is selective partial pancreatectomy, while diffuse CHI necessitates long-term drug therapy or subtotal pancreatectomy [3,6].
To date, underlying genetic mutations for CHI are identified in 45%â55% of cases. Fourteen genes have been identified (ABCC8, KCNJ11, GLUD1, GCK, HADH, HNF1A, HNF4A, SLC16A1, UCP2 and more recently CACNAID, FOXA2, HK1, PGM1, and PMM2) [7]. That approximately half of the cases do not have an identifiable genetic etiology suggests that additional loci are undiscovered [8,9]. Notably, CHI most frequently results from mutations in KATP-channel genes, ABCC8 or KCNJ11 [2,3,9,10]. Diazoxide-unresponsive CHI can be caused by homozygous or heterozygous KATP-channel mutations leading to diffuse CHI. Alternatively, a paternal KATP-channel mutation and somatic loss of heterozygosity of chromosome 11p15 lead to focal diazoxide-unresponsive CHI. CHI can be nonsyndromic or associated with a range of syndromes, including Beckwith-Wiedemann, Turner, and Kabuki syndrome [11]. Novel manifestations in apparently non-syndromic CHI patients can represent a novel syndromic association, unrecognized medical side effects, or chance findings [10]. Notably, spontaneous CHI remission and reversion to diabetes in adulthood have been reported in patients harboring mutations in ABCC8, HNF4A, or HNF1A [12]. Many patients with monogenic diabetes can be effectively treated with oral antidiabetic medication instead of insulin, which underscores the importance of a genetic diagnosis [13].
According to recent American College of Medical Genetics guidelines [14], novel changes should be considered variants of unknown significance (VUS) and should not be considered pathogenic until the variant is verified in other patients. Accordingly, identifying novel variants associated with reversal of CHI to diabetes in adult life has implications for genetic cascade screening of family members.
We describe 2 patients with novel ABCC8 mutations associated with spontaneous clinical remission.
Case reports
1. Case 1
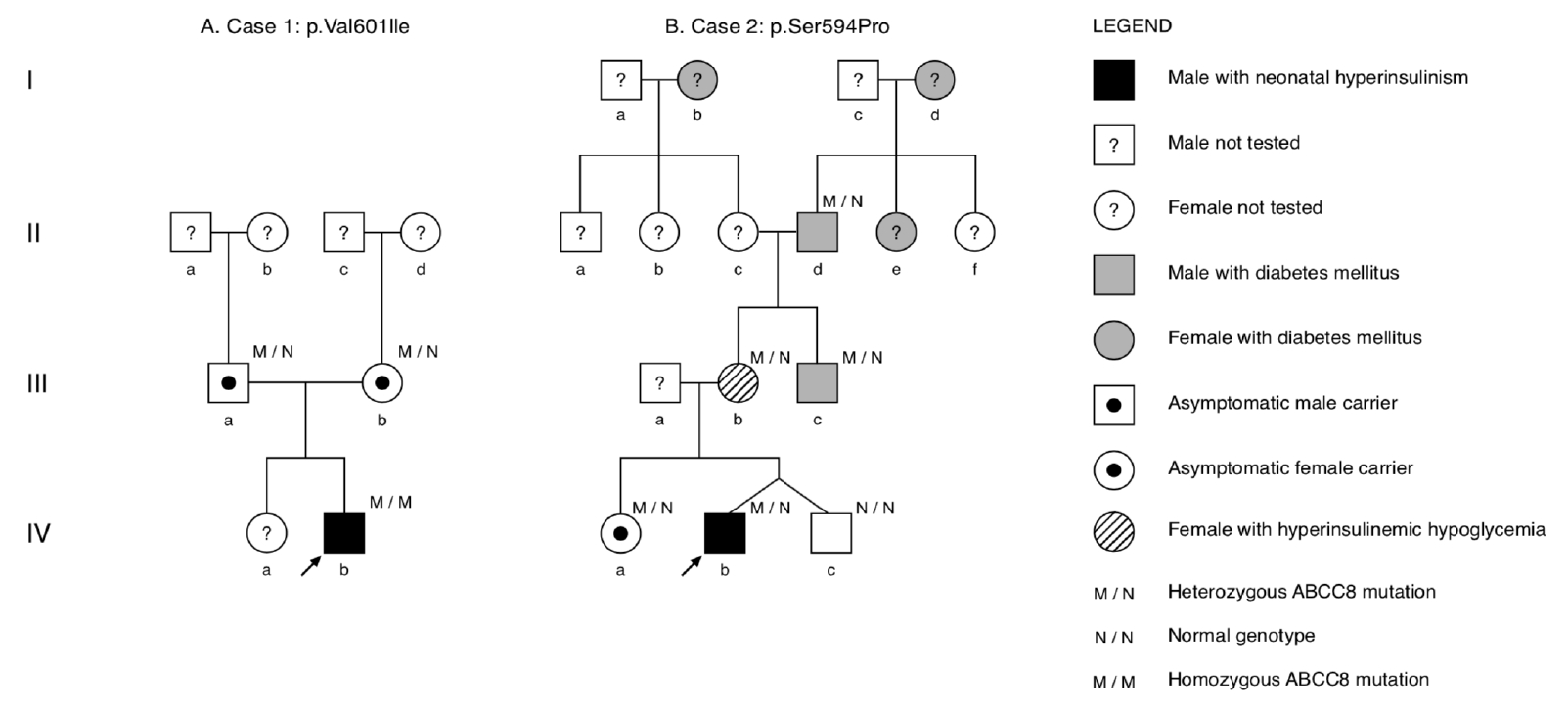
A Caucasian male patient was born via normal vaginal delivery at 331/7 weeks gestation to nonconsanguineous parents (Fig. 1A). The pregnancy was unremarkable with no gestational diabetes mellitus. The patient was large-for-gestational age (birthweight, 3,150 g; length, 51 cm; both >97th percentile) with no dysmorphic features. Apgar scores were 5, 8, and 8 (at 1, 5, and 10 minutes, respectively). At 2 hours of life, he developed severe symptomatic hypoglycemia (blood glucose <0.1 mmol/L) with hypotonia. A critical blood sample was drawn, and intravenous (IV) glucose infusion (up to 19 mg/kg/min) was initiated to maintain normoglycemia.
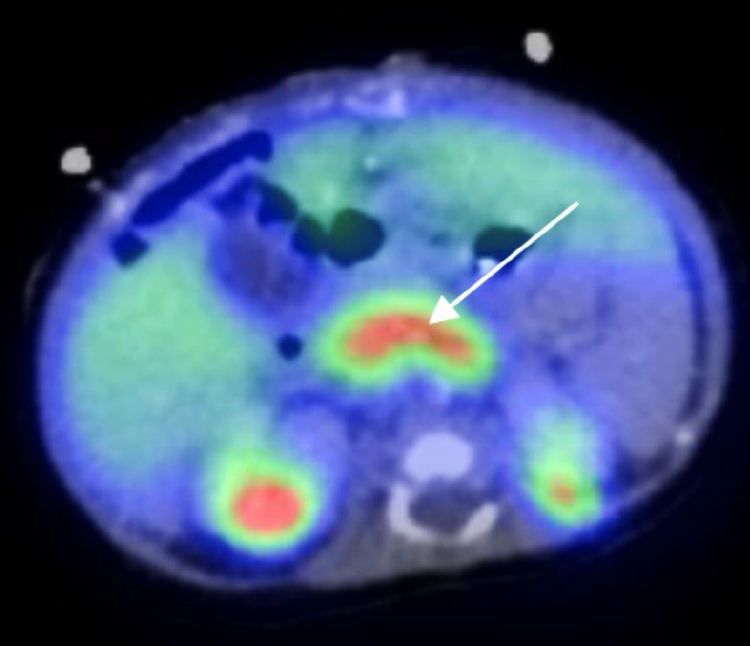
During hypoglycemia, serum insulin, and C-peptide levels were elevated in the setting of suppressed ÎČ-hydroxybutyrate and free fatty acids (Table 1). Cortisol, growth hormone, and thyroid hormones were within normal limits, and metabolic and infection screening were negative. Analysis with 18F-dihydroxyphenylalanine positron emission tomography-computed tomography (Discovery 690, GE Healthcare, Chicago, IL, USA) showed general uptake of glucose from the pancreas, suggestive of diffuse CHI (Fig. 2). Oral diazoxide (10 mg/kg/day) was initiated on day 7 of life. Diazoxide combined with high-rate IV glucose infusion (12 mg/kg/min) maintained blood glucose level within normal range. Oral feeding was difficult, and the child developed emesis. Intestinal malrotation, and volvulus of the ileum were diagnosed on day 9 of life. Diazoxide was discontinued, and 76 cm of necrotic ileum/jejunum was resected with intestinal stoma. Octreotide (up to 48 ”g/kg/day subcutaneous, continuously) and amlodipine (0.1 mg/kg/day) were introduced 7 days after surgery, which stabilized blood glucose level with a persistent need for IV glucose in the high-normal range (6â7 mg/kg/min).
Molecular analysis (International Clinical Laboratory of University of Exeter, Exeter,UK) revealed that the proband harbored a homozygous missense DNA variant, c.1801G>A, p.(Val601Ile), in exon 12 of ABCC8 (reference sequence: NM_001287174.1) (Fig. 3). Amino acids in this residue are evolutionary conserved. Both PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and PANTHER (http://pantherdb.org/tools/csnpScoreForm.jsp) in silico algorithms predicted the rare variant to be pathogenic (Table 1).
The unaffected parents were both heterozygous carriers of the ABCC8 variant. To our knowledge, only 1 case harboring this mutation has been reported [15]. In this report, the ABCC8 mutation in the male newborn was paternally inherited, and diazoxide was discontinued after 14 months. In the current case, octreotide was weaned after 3 days, and diazoxide treatment was reintroduced followed by a progressive decrease in IV glucose infusion, which was finally stopped at day 54 of life. The intestinal stoma was reversed at 67 days of life with no evidence of short bowel syndrome.
At follow-up at 2 years of life, the patient had no hypoglycemic episodes, and diazoxide was discontinued. A subsequent fasting tolerance test was age-appropriate, and he exhibited normal growth and neurological development.
2. Case 2
A Hispanic male patient and his healthy, dizygotic twin brother were born via emergency Caesarian section at 344/7 weeks gestation due to abnormal fetal heart rate on cardiotocographic monitoring. The pregnancy was uneventful without gestational diabetes, and no formal glucose tolerance testing was performed. The nonconsanguineous parents had a healthy 3-year-old daughter, and family history revealed several maternal family members with diabetes mellitus (Fig. 1B).
The newborn infant had a birth weight of 2,150 g (50th percentile), while his twin brother weighed 2,550 g. The patient had moderate asphyxia and required noninvasive ventilator support for several days. On his first day of life, he experienced severe hypoglycemia (blood glucose<0.1 mmol/L) and required IV glucose (up to 21 mg/kg/min) to maintain normoglycemia. Physical examination was unremarkable. The diagnosis of CHI was based on hypoglycemic episodes (<2.4 mmol/L) on the first day of life, unsuppressed insulin level during hypoglycemia, and high carbohydrate requirements to maintain normal glycemic level (Table 1). Diazoxide treatment promptly resolved the hypoglycemia.
Molecular testing (Odense University Hospital, Odense, Denmark) identified a novel, heterozygous DNA variant, c.1780T>C, p.(Ser594Pro), in exon 12 of ABCC8 (reference sequence: NM_000352) (Fig. 3). The residue is evolutionarily conserved, and 1 of 2 in silico algorithms predicted the variant to be likely deleterious (Table 1). Based on the American College of Medical Genetics guidelines, we considered the change to be a VUS. Diazoxide was discontinued at 15 months of age. Follow-up at 7 years of age revealed normal growth and psychomotor development. He had age-appropriate fasting tolerance without hypoglycemic events. However, a subsequent oral glucose tolerance test (OGTT) revealed moderate hyperinsulinism (+30 minutes: insulin, 58.8 ”U/L [normal range, 30â50 mUl/L]; glucose, 7.7 mmol/L) followed by marginal, nonsymptomatic hypoglycemia at +120 minutes (insulin, 7.37 ”U/L; glucose, 3.1 mmol/L) (Table 2).
Interestingly, the mother (Fig. 1B, lllb) reported frequent episodes of lightheadedness. Subsequently, an OGTT revealed hyperinsulinism in the mother at +30 minutes (185.5 mU/L) that induced symptomatic HH at +120 minutes (blood glucose, 3 mmol/L; insulin, 27 mU/L) (Table 2). The proband's maternal uncle (Fig. 1B, IIIc) had diabetes mellitus, which was being treated with insulin. Lab testing was negative for antiglutamic acid decarboxylase, anti-insulin, and anti-IA2 antibodies, and serum C-peptide level was 0.68 ”g/L, which was only slightly below the normal range (1.0â3.1 ”g/L). On insulin therapy, the uncle had good metabolic control (glycated hemoglobin) yet suffered frequent hypoglycemic episodes. The maternal great-grandmother (Fig. 1B, Ib) also had been diagnosed with diabetes mellitus, but no detailed clinical information was available.
Cascade screening in the family revealed that the probandâs older sister (Fig. 1B, IVa), mother (Fig. 1B, lllb), maternal uncle (Fig. 1B, lllc), and maternal grandfather (Fig. 1B, IId) harbored the same heterozygous ABCC8 variant p.(Ser594Pro). Consequent to genetic testing, the probandâs maternal uncle was rediagnosed with monogenic ABCC8-diabetes. He was transitioned from insulin to oral antidiabetic medication (glibenclamide 3Ă5 mg daily) and experienced no further hypoglycemic episodes.
Discussion
We report 2 patients with CHI caused by novel DNA variants in ABCC8. Patient 1 was diagnosed with CHI due to a homozygous ABCC8 mutation inherited from unaffected parents, which was consistent with an autosomal recessive mode of transmission. The patient was unresponsive to diazoxide treatment, and oral intake was not possible. Subtotal pancreatectomy was considered as a potential treatment option; however, a literature review revealed a case of a patient that harbored the identical missense mutation that had responded to diazoxide treatment [15]. Based on the single case observation, we opted to postpone pancreatectomy and use alternative treatment (octreotide and amlodipine) during the postoperative fasting period prior to resuming oral diazoxide. Thus, detailed clinical investigation and scholarly inquiry led to an appropriate clinical approach and avoided irreversible surgical intervention. Cochrane and GRADE criteria consider case reports to be weak levels of evidence. However, for the present case, this information was the best available evidence and helped guide the therapeutic approach [6]. This example demonstrates how case reports can inform clinical care, particularly in the setting of rare diseases such as CHI.
Intestinal malrotation occurs in approximately 1 in 500 living births. Malrotation is estimated to be symptomatic 1 in 6,000 live births, and up to 80% of symptomatic cases occur in newborns [16]. Risk factors for volvulus include birth defects such as intestinal malrotation, Hirschsprung disease, and isomerism or heterotaxy syndrome. Recently, rare copy number variations (CNVs) have been identified in patients with syndromic intestinal malrotation, suggesting that CNV screening could be informative in cases when intestinal malrotation occurs in a constellation of other malformations [17]. To the best of our knowledge, this is the first report that associated congenital malrotation with neonatal hyperinsulinism. A recent study found 'likely pathogenic' or 'pathogenic' CNVs in 4 of 47 patients (8.5%) with intestinal malrotation [17]. To date, no reports have implicated the 11p15.1 region of ABCC8 in intestinal malrotation. We are not aware of any study that has identified an association between diazoxide therapy with volvulus, and this possibility merits consideration. It is unclear if co-occurrence of malrotation and CHI is merely a coincidence or if there is a true causal association. Therefore, studies in larger patient cohorts are needed to clarify the potential link between ABCC8 mutations and midgut malrotation.
Case 2 documents transient CHI due to a heterozygous mutation in ABCC8, which is consistent with an autosomal dominant (AD) mode of transmission. A detailed, multigenerational family histor y and cascade screening facilitated reclassification of the maternal uncle's diabetes mellitus. Patients with monogenic diabetes can be treated with oral antidiabetic medications instead of insulin [13]. Thus, molecular analysis enabled treatment modification from insulin to oral antidiabetic medication.
As reported by Kapoor and colleagues, siblings harboring the identical AD mutation in ABCC8 exhibit variable phenotypes, which can range from asymptomatic macrosomia to persistent HH in childhood to glucose intolerance or early-onset diabetes mellitus in adulthood [18]. Vieira et al. [19] described a patient carrying a de novo AD mutation in ABCC8 in which HH evolved to gestational diabetes and subsequently into diabetes mellitus.
Gain-of-function mutations in ABCC8 or KCNJ11 lead to KATP-channel hyperactivity causing neonatal diabetes. Conversely, loss-of-function mutations that reduce channel activity cause CHI. These observations can be explained by the variable affinity of mutant sulfonylurea receptors for adenosine triphosphate magnesium salt [20]. However, it remains unclear how the identical mutation can be associated with disparate phenotypes (i.e., CHI and diabetes) within the same pedigree. Some have hypothesized that the primary insulin secretion defect relates to chronically increased intracellular calcium level that activates apoptotic pathways [18].
In the case in this study, the patient and his unaffected older sister (Fig. 1B, IVa) might have increased risk for developing diabetes mellitus later in life. Accordingly, careful long-term medical follow-up is warranted. The genotype-phenotype correlations in pedigree 2 should be interpreted with caution as other family members with diabetes mellitus did not undergo genetic testing. Although clinical evidence supports the pathogenicity of the identified novel mutation, to-date, in silico analyses showed discrepant results, and in vitro functional studies of the identified ABCC8 mutants have not been conducted.
In conclusion, the 2 case reports reported herein illustrate highly variable phenotypes in patients harboring mutations in ABCC8. We report a unique finding of intestinal malrotation and volvulus that occurred 2 days after diazoxide treatment in a patient with CHI harboring a homozygous ABCC8 mutation. Further studies are needed to determine if these entities are pathophysiologically related. The cases demonstrate the importance and clinical utility of genetic analyses for informing and guiding treatment and care. First, only rare cases (case 1) of autosomal recessive, biallelic ABCC8 mutations are diazoxide-responsive. Second, genetic analysis should be performed in all patients that present with CHI to define the best treatment approaches. Finally, patients with heterozygous mutations in ABCC8 and their family members should be closely followed clinically, as they might be at increased risk for developing diabetes mellitus.