Introduction
Cushing syndrome (CS) describes clinical manifestations due to chronic exposure to excess glucocorticoids [1]. The most common cause of CS is iatrogenic [1], and endogenous CS occurs rarely, with an overall incidence of 0.7 to 2.4 cases per million people per year [2]. Among the new cases of endogenous CS, only about 10% occur in children [3,4]. CS is classified into two types, adrenocorticotropic hormone (ACTH)-dependent and ACTH-independent CS. Cushing disease (CD), overproduction of ACTH by a pituitary tumor, is the most common cause of ACTH-dependent CS [4]. ACTH-independent CS, including adrenal adenoma, adrenal carcinoma, and micronodular adrenal hyperplasia, accounts for approximately 15% of CS in childhood [3]. Adrenocortical tumors are rare in children and account for approximately 0.2% of all pediatric neoplasm [5]. Moreover, cortisol-secreting adrenal adenomas are even more rare [6].
The clinical presentation of CS varies according to extent and duration of glucocorticoid excess [3,4]. Urolithiasis is a rare disease in children [7], but one of the common complications of CS [8]. Although urolithiasis is more common in children with CS than in those without, it is usually underrecognized because of its rarity [7,9]. Early recognition of urolithiasis is important in pediatric CS, particularly with respect to treatment outcomes. Early diagnosis of urolithiasis allows conservative management or stone removal without complications. In contrast, late diagnosis can be followed by serious complications, including urinary tract infection (UTI), sepsis, hematuria, and acute kidney injury (AKI) [9,10].
Here, we report the first case of a patient with CS associated with AKI due to urolithiasis. This 6-year-old boy was treated with etomidate for hypercortisolism and continuous renal replacement therapy (CRRT) for renal function, followed by surgery for an adrenal adenoma and ureteral stones.
Case report
A 6-year-old boy was referred to our center for evaluation of an adrenal mass. Prior to transfer, he visited the Emergency Department of another hospital with the chief complaint of generalized tonic-clonic type seizure. He was diagnosed with posterior reversible encephalopathy syndrome due to hypertension based on brain computed tomography (CT) and magnetic resonance imaging (MRI). Abdominal CT for evaluation of secondary hypertension showed an adrenal mass suggestive of pheochromocytoma. The patient was treated with oral nifedipine (Adalat OROS, Bayer Pharmaceuticals, Newbury, UK), intravenous (IV) labetalol HCl (Labesin, Myungmoon Pharmacueticals, Seoul, Korea), and IV dexamethasone for hypertensive encephalopathy and transferred to our center for further evaluation and surgical resection of the mass.
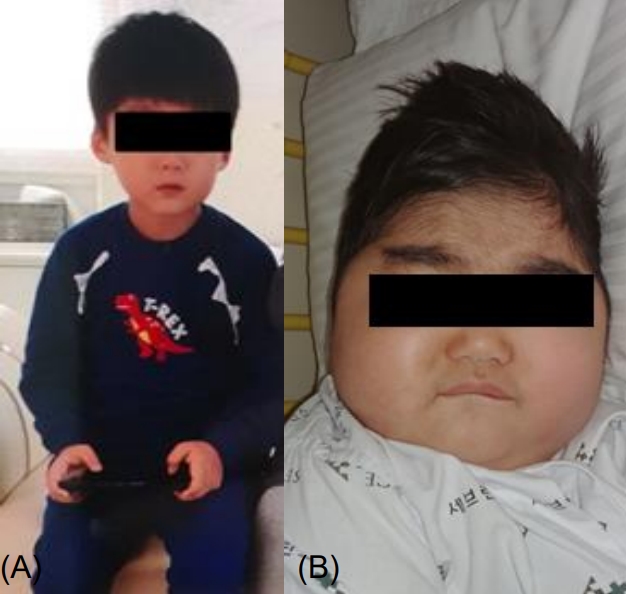
The patient was born at 40 weeks of gestation and had no remarkable perinatal complications or medical and treatment history. Family history was significant for hyperthyroidism in his mother and thyroid cancer in his grandfather. He had no symptoms of headache, palpitation, or diaphoresis. He had gained 20 kg of weight in the past 10 months. The patientŌĆÖ vital signs were as follows: pulse rate, 110 beats/min; blood pressure, 170/110 mmHg (>99th percentile); respiratory rate, 18 times/min; and body temperature, 36.6Ōäā. On physical examination, the patient had moon face, central obesity, Buffalo hump, abdominal striae, and hirsutism (Fig. 1). His anthropometric measurements were as height; 125 cm (1.21 height standard deviation score [SDS]) weight; 35 kg (2.54 weight SDS), body mass index; 23.04 kg/m2 (>97th percentile).
On laboratory tests, complete blood counts, electrolytes, liver function, and renal functions tests were normal. Fasting glucose was 96 mg/dL (normal <100 mg/dL), and hemoglobin A1c level was 5.7% (normal, 4.0%ŌĆō6.0%). Urine analysis showed proteinuria and glycosuria, and the urine calcium/creatinine ratio was 0.23 (normal, <0.14). Electrocardiogram, echocardiography, and kidney doppler ultrasonography findings were normal.
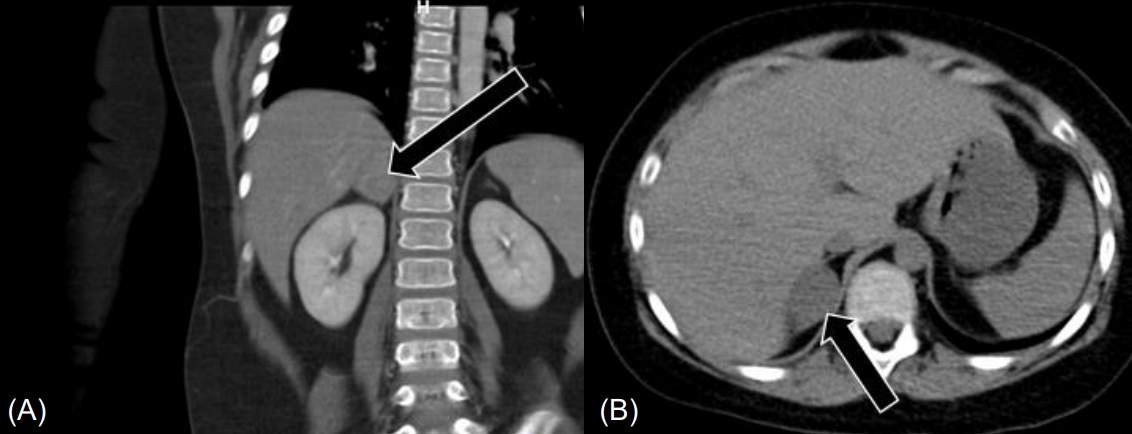
Abdominal CT showed a 2.7-cm-sized heterogeneously enhanced mass lesion with a CT attenuation value of 25.1 Hounsfield units (HU) in the right adrenal gland, suggesting pheochromocytoma, and a tiny stone in the right distal ureter (Fig. 2). Abdominal MRI showed a 3.3-cm-sized enhancing mass in the right adrenal gland without signal drop in the fat tissue on the opposed phase, also suggesting pheochromocytoma.
Endocrinologic tests suggested ACTH-independent CS due to adrenocortical tumor with the following findings: elevated 24-hour urinary free cortisol (UFC), 1529.2 ╬╝g/day (normal, 58.0ŌĆō403.0 ╬╝g/day); elevated midnight serum cortisol level, 26.4 ╬╝g/dL (normal, <2.0 ╬╝g/dL); and unsuppressed serum cortisol level after low-dose dexamethasone suppression test (DST), 30.9 ╬╝g/dL (normal, <5.0 ╬╝g/dL). As the plasma ACTH level at midnight and 8:00 AM were undetectable, at less than 1.5 pg/mL (normal, 7.2ŌĆō63.3 pg/mL), and an adrenal mass was detected in abdominal CT and MRI, high-dose DST was not performed. Primary aldosteronism and pheochromocytoma were ruled out with the following findings: aldosterone/renin ratio, 11 ng/dL per ng/mL/hr (normal <20 ng/dL per ng/mL/hr); plasma metanephrine, 0.07 nmol/L (normal, 0ŌĆō0.50 nmol/L); plasma normetanephrine, 0.09 nmol/L (normal, 0ŌĆō0.09 nmol/L); 24-hour urine metanephrine, 65.9 ╬╝g/day (normal, 52ŌĆō341 ╬╝g/day); and 24-hour urine normetanephrine, 86.9 ╬╝g/day (normal, 88.0ŌĆō444.0 ╬╝g/day).
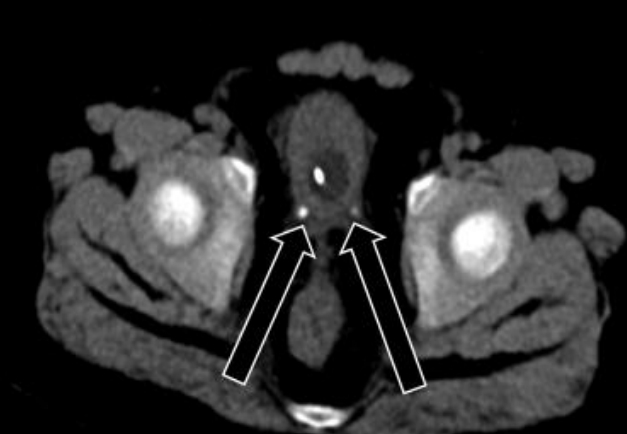
While waiting for surgery, the patient complained of abdominal pain, hematuria, and anuria. Abdominal CT was performed again and showed bilateral distal ureter stones without changes in the adrenal mass (Fig. 3). The patient started getting drowsy, and his blood pressure was not well-controlled despite the maximum doses of oral losartan K (Cozaar, MSD-Korea, Seoul, Korea), spironolactone (Aldactone, Pfizer AG, Zrich, Schweiz), minoxidil (Minoxidil, Hyundai PharmCo. Ltd., Seoul, Korea), and I V nicardipine (Perdipine, Dong-A Pharm., Seoul, Korea). Blood urea nitrogen and creatinine levels were elevated to 41.5 mg/dL (normal, 8.0ŌĆō18.5 mg/dL) and 2.32 mg/dL (normal, 0.32ŌĆō0.70 mg/dL), respectively. Because his condition deteriorated rapidly with AKI, he was transferred to the intensive care unit (ICU) and underwent CRRT before preparation of ureteral stent indwelling. As fever developed and his C-reactive protein level was elevated to 135.1 mg/L (normal, 0ŌĆō8.0 mg/L), broad-spectrum antibiotics were administered. To control hypercortisolism, we started IV etomidate infusion at a rate of 0.03 mg/kg/hr. Serum cortisol level was assessed every 4ŌĆō6 hours, and the dose of etomidate was decreased to 0.02 mg/kg/hr to normalize the serum cortisol level. When the serum cortisol level reached 5.5 ╬╝g/dL (normal, 6.0ŌĆō18.4 ╬╝g/dL), IV hydrocortisone infusion at a rate of 15 mg/m2/24 hr was started and used to maintain the serum cortisol level between 8.7 ╬╝g/dL and 12.4 ╬╝g/dL. After maintaining a normal cortisol level and performing ureteral stent indwelling, the patientŌĆÖs blood pressure improved, and losartan K, minoxidil, and nicardipine were discontinued. After confirming improvement of oliguria and renal function, CRRT was discontinued. In preparation for resection of the adrenal mass, IV etomidate infusion was discontinued 1 hour before the surgery, and IV hydrocortisone was increased to 100 mg/m2/24 hr. The patient underwent laparotomy and resection of a well-encapsulated right adrenal mass, which was histologically confirmed as an adrenocortical adenoma. The surgery was successfully performed without perioperative complications. Postoperatively, his condition was significantly improved, and IV hydrocortisone was changed to oral medication. The ureteral stones were removed, and analysis showed calcium carbonate apatite. The patient was discharged with replacement therapy of hydrocortisone at 15 mg/m2/day.
Discussion
Our patient showed seizure due to hypertensive encephalopathy and AKI due to bilateral ureteral stones, as well as common features of CS such as rapid weight gain, moon face, central obesity, Buffalo hump, and purple striae. Although a case of hypertensive encephalopathy associated with CS has been reported [11], to our knowledge, there is no reported case of CS with AKI due to urolithiasis.
Urolithiasis is a rare disease in children, with an annual incidence of 36 to 57 per 100,000 children in the United States [9]. Urolithiasis in children is more common in those with risk factors such as urinary tract anomaly or infection, neurogenic bladder, tubular disorder, genetic disorder, or systemic disease [7]. In these patients, excessive solutes including calcium, oxalate, phosphate, citrate, uric acid, and cysteine in the urine and their crystallization promote stone formation [9]. Rahman et al. [8] reported that kidney stones were more common in children with CD than in those without. Obesity, insulin resistance, hypertension, and dyslipidemia, as well as hypercalciuria, hyperuricosuria, and hypercystinuria induced by hypercortisolism increase the risk of urolithiasis in CS [8]. Our patient showed obesity, hypertension, dyslipidemia, and hypercalciuria. In patients with these risk factors, careful monitoring for urolithiasis is required. The calcium carbonate apatite found in our patient's stone analysis is associated with UTI. Carbon dioxide, formed by breakdown of urea by urease produced by bacteria, breaks down to carbonate, which combines with calcium and phosphate to form a calcium carbonate apatite stone. This stone is usually asymptomatic or manifests as fever, flank, or abdominal pain, hematuria, UTI, sepsis, and/or renal insufficiency. The standard diagnostic modality for such stones is CT, and broad-spectrum antibiotics with emergency decompression are required for improving obstructive pyelonephritis. Although older studies have proposed withholding emergency decompression in patients with pyelonephritis [12], prompt decompression via ureteral stent insertion or nephrostomy tube now is preferred for better prognosis [13]. Although a unilateral ureteral stone was observed on initial abdominal CT, our patient had no symptoms indicating urolithiasis. However, fever, abdominal pain, hematuria, and anuria occurred later due to bilateral ureteral obstruction. In patients with CS accompanied by these symptoms, proper evaluation of urolithiasis is required.
In this case, AKI occurred due to bilateral ureteral obstructions by stones. The etiology of AKI is classified into prerenal, intrinsic renal, and postrenal, with intrinsic renal cause being the most frequent (63.8%), followed by postrenal (24.1%.) and prerenal cause (9.5%). Among postrenal causes of AKI, obstructive urolithiasis is the most common (85.7%) [14]. Although AKI is associated with longer ICU stay and mechanical ventilation, higher mortality rates, and chronic kidney disease (CKD), no single treatment modality has been shown to reverse AKI, and renal replacement therapy often is required [15]. Consequently, careful monitoring and management of AKI etiologies are required for hospitalized children. In patients with CS, although CKD with a decreased glomerular filtration rate is caused by chronic glucocorticoid exposure [16], there are no studies on the relationship between CS and AKI.
In investigation of a patient with suspected CS, step-by-step biochemical laboratory tests are required. Loss of circadian rhythm of plasma cortisol, elevated midnight salivary cortisol, increased UFC excretion, and unsuppressed cortisol response after overnight/low-dose DST are positive findings for CS diagnosis. After confirming CS, subsequent evaluations including midnight ACTH, high-dose DST, corticotropin-releasing factor test, and inferior petrosal sinus sampling should be performed to determine the cause of CS, if indicated. Imaging studies including CT/MRI scanning of pituitary and adrenals and scintigraphy studies are required for CS localization [1,3,4]. Although the radiological findings on abdominal CT and MRI suggested pheochromocytoma, our case was diagnosed as CS by clinical manifestations, biochemical studies including elevated UFC, and unsuppressed cortisol level after low-dose DST; whereas a normal range of catecholamine was noted in urine and serum. Furthermore, adrenocortical adenoma was confirmed upon pathological examination. In general, adrenal adenomas contain abundant intracellular lipids, which can easily be distinguished from pheochromocytoma through CT or MRI. As intracellular lipids of the adrenal adenoma reduce CT attenuation, attenuation of lipid-rich adenoma is less than 10 HU [17]. However, lipid-poor adenoma is difficult to distinguish from pheochromocytoma through CT or MRI because of insufficient intracellular lipids [17,18]. Due to overlap of clinical manifestations between adrenal adenoma and pheochromocytoma, the possibility of lipid-poor adrenal adenoma should be considered in cases with both imaging findings of pheochromocytoma and clinical manifestations of CS.
In our patient, IV etomidate was administered to control hypercortisolism as resection of the adrenal mass was delayed due to deteriorating condition. Etomidate, a carboxylated imidazole, blocks cortisol production by inhibiting the activity of the adrenal enzyme 11╬▓-hydroxylase, which converts deoxycortisol into cortisol and can be used for severe hypercortisolism before CS surgery to control complications of hypercortisolism [19]. Although there is little pediatric experience with etomidate, Greening et al. [20] reported a case of pediatric CD treated with IV etomidate at a rate of 0.03 mg/kg/hr prior to surgical cure.
Herein, we report the first case of CS with AKI caused by urolithiasis. Variable risk factors were associated with bilateral ureteral stones in our patient and caused deteriorated condition and AKI. This case demonstrates that patients with CS require attentive evaluation and proper management of urolithiasis as one of the clinical manifestations of CS.