Highlights
We conducted a cell function study for a novel variant (p.Tyr825Phe[Y825F]) of the CASR gene in a neonate with congenital hypoparathyroidism and hypercalciuria. This proved that the CASR-Y825F expressed in cells elevated MAPK signaling and increased [Ca2+]i to induce hypocalcemia.
Introduction
Autosomal dominant hypocalcemia with hypercalciuria (ADHH) is a genetic disorder characterized by hypocalcemia and hypercalciuria with inappropriately low levels of parathyroid hormone (PTH) [1]. Patients with ADHH exhibit varying degrees of hypocalcemia [2]. In addition to hypocalcemia, hypomagnesemia, and hypercalciuria, Bartter syndrome may also occur.
which is located on chromosome 3q13.3–21 and consists of 6 exons [3,4]. CASR is composed of 1,078 amino acids and contains a large extracellular domain comprising 7 transmembrane (TM)-spanning domains and an intracellular tail [5]. CASR is a cell membrane G protein–coupled receptor that is generally located in the parathyroid glands and renal tubules; this receptor detects minute changes in serum calcium levels and is responsible for the regulation of parathyroid secretions and positive ions in the kidneys [6,7]. CASR maintains calcium homeostasis by regulating extracellular calcium ion ([Ca2+]e) levels through the detection of intracellular calcium ion ([Ca2+]i) levels. There is an inverse relationship between PTH secretion and [Ca2+]e concentration, and a disruption of calcium homeostasis leads to complications, such as hypercalcemia or hypocalcemia [8]. In patients with chronic hypocalcemia, CASR increases the expression of the PTH gene and stimulates parathyroid cell proliferation. In contrast, the synthesis and secretion of PTH are suppressed when the blood calcium concentration is high. As such, inactivating or activating CASR mutations can cause hyperparathyroidism or hypoparathyroidism [9].
Activating mutations of CASR, or gain-of-function CASR mutations, have been shown to result in ADHH, and various CASR mutations have been reported. We previously reported the first case of a novel activating variant of CASR (Y825F) that occurred de novo in a neonate with hypoparathyroidism, hypercalciuria, and hypomagnesemia [10-13]. Y825F is located in the TM domain of CASR, and previous studies reported pathogenic activating variants of A824P and I816T, which are also located in the TM domain [5].
A patient with a history of convulsions was transferred to the neonatal intensive care unit at the Kyungpook National University Children's Hospital. At the time of admission, the serum calcium level was 4.7 mg/dL (9.0–10.6 mg/dL), ionized calcium level was 2.2 mg/dL (4.8–4.92 mg/dL), phosphate level was 13.2 mg/dL (4.8–8.2 mg/dL), magnesium level was 1.29 mEq/L (1.44–3.12 mEq/L), PTH level was 2.6 pg/mL (10–65 pg/mL), and vitamin D level was 13.95 ng/mL (8.0–51.9 ng/mL). The patient did not have any morphological abnormalities of the face, limbs, or fingers. Cardiac ultrasonography revealed a 3.4-mm atrial septal defect, but the renal ultrasonogram was normal. The patient's thymus gland was normal, as revealed by chest ultrasonography, and the test for 22q11.2 deletion syndrome was negative. In blood tests repeated during the follow-up period, inappropriately low PTH levels (intact-PTH, 0–6 pg/mL) were observed despite the hypocalcemia. The urine calcium/urinary creatinine ratio was elevated (2.0). The patient was shown to carry a novel CASR heterozygous variant in exon 7 (c.2474A>T; p.Tyr825Phe). The variant was predicted to be deleterious, with a Sorting Intolerant From Tolerant score of 0, and potentially deleterious, with a Polyphen-2 HVAR score of 0.994. Additionally, the variant was predicted to be a likely pathogenic variant as the associated Phastcon and Genomic Evolutionary Rate Profiling scores were high (0.994 and 5.89, respectively).
In this study, we conducted in vitro functional analyses to determine whether the novel CASR variant (Y825F) found in the neonate with hypocalcemia and hypercalciuria was disease-causing by evaluating calcium regulation and changes in intracellular signaling in kidney cells expressing this variant.
Materials and methods
Reagents related to cell culture were purchased from Gibco (Grand Island, NY, USA). Minimum essential medium (MEM) and Dulbecco's Modified Eagles' Medium (DMEM, high glucose) were purchased from Welgene (Gyeongsan, Korea), and DMEM (high glucose, (-) calcium chloride) and opti-MEM were purchased from Gibco (Grand Island). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA, USA). Other reagents used in the study were purchased from Sigma-Aldrich (St. Louis, MO, USA).
1. Preparation of expression vectors
RNA was extracted from human pancreatic cells using the RNeasy Mini kit (QIAGEN, Hilden, Germany) and cDNA was synthesized using the SuperScript III First-Strand synthesis system (Invitrogen). We amplified wild-type (WT) CASR (NCBI accession No. NM_000388.3) by polymerase chain reaction (PCR) using PrimeSTAR Max DNA Polymerase (Takara Bio, Kusatsu, Japan). PCR was conducted according to the following reaction conditions: initial denaturation at 95℃ for 5 minutes, followed by 35 cycles of denaturation at 95℃ for 30 seconds, annealing at 58℃ for 30 seconds, and extension at 72℃ for 7 minutes, and a final extension at 72℃ for 10 minutes. The amplified PCR product was digested with HindIII and NheI restriction enzymes and cloned into the pSF-CAG-Amp (Oxford Genetics, Oxford, UK) vector. We performed mutagenesis on the WT CASR vector using PrimeSTAR Max DNA Polymerase and specific primers. The resulting products were treated with Dpn1 (Takara Bio) and transformed into competent cells. Plasmids were isolated from selected colonies using the plasmid mini kit (GeneAll Biotechnology, Seoul, Korea). The WT CASR expression vector (CASR-WT) and mutant CASR expression vector (CASR-Y825F) were confirmed by sequencing. Plasmid preparation for transfection was performed using the Nucleo-Bond Xtra Midi kit (MACHEREY-NAGEL, Düren, Germany). The primers used for vector preparation are shown in Table 1.
2. Cell culture and transient transfection
HEK293 cells derived from human embryonic kidney epithelial cells (KCLB No. 21573) were provided by the Korea Cell Line Bank. The cells were maintained in MEM supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin (1×PS) at 37℃ in a 5% CO2 incubator. The medium was replaced with fresh medium every 2 days.
Lipofectamine 2000 was used to transfect HEK293 cells with the CASR-WT or CASR-Y825F vector. Cells were seeded in 6-well plates (6×105 cells per well) and cultured for 2 days. Transfection reactions were prepared by mixing opti-MEM, 4 μg expression vector, and 2.5× lipofectamine and allowing it to react at room temperature for 20 minutes; the reactions were then added to cells following the manufacturer's requirements. After 12 hours of incubation in a 5% CO2 incubator at 37℃, the medium was replaced with DMEM containing 10% FBS and 1×PS. At 48-hour posttransfection, the medium was removed; the cells were washed once with Dulbecco's phosphate-buffered saline (DPBS) (Welgene) and used for experiments.
3. Western blot analysis
1) Determination of CASR protein expression
Cells transfected with CASR-WT or CASR-Y825F were lysed in radio immunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin) containing 1 mM phenylmethanesulfonyl fluoride (Cell Signaling Technology, Inc., Danvers, MA, USA), and protein concentrations of cell lysates were quantified with the 4Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). To evaluate dimer formation, we used 5× sample buffer without β-mercaptoethanol (Biosesang, Seongnam, Korea). Protein samples (50 μg) were separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) at 80–120 V and then transferred onto a polyvinylidene fluoride (PVDF) membrane (Merck-Millipore, Darmstadt, Germany) for 16 hours (or overnight) at 50 V using a transfer device. To confirm protein transfer, membranes were stained with Ponceau S (Sigma-Aldrich), centrifuged at 13,000 rpm for 30 minutes at 4℃, and the resultant supernatant was isolated. Proteins of the supernatant were stained using the bicinchoninic acid (BCA) method (Pierce BCA) and washed 3 times with TBST buffer (Tris-buffered saline with 0.1% Tween 20) for 10 minutes. Membranes were blocked with 5% skim milk for 1 hour and then incubated with primary antibody, anti-CASR monoclonal antibody (sc-47741, Santa Cruz Biotechnology, Inc., Dallas, TX, USA), overnight at 4℃. After washing the membrane 5 times with TBST for 10 minutes each, the membranes were incubated with the secondary antibody, anti-mouse-IgG-HRP (#7076, Santa Cruz Biotechnology, Inc.), for 1 hour at room temperature. After washing the membrane 5 times with TBST for 10 minutes, the membrane was processed using enhanced chemiluminescence (iNtRON Biotechnology, Seongnam, Korea), and the bands were quantified using ImageQuant LAS 4000 (GE Healthcare, Chicago, IL, USA).
2) Determination of MAPK activation
To determine mitogen-activated protein kinase (MAPK) activation in transfected cells, the medium was replaced at 24-hour posttransfection with DMEM ((-)calcium chloride) containing 0.1% FBS and serum starvation was induced for 24 hours. The cells were then treated with 0, 2, 4, or 10 mM calcium solution (#21115, Sigma-Aldrich) for 10 minutes. The calcium-containing medium was quickly removed, and the cells were washed with DPBS. Cells were lysed using RIPA buffer as described above and protein concentrations were quantified. Protein samples (15–20 μg) were mixed with sample buffer, separated by 12% SDS-PAGE on a 12% acrylamide gel, and transferred onto a PVDF membrane. The membranes were first probed with phospho-specific primary antibodies and then stripped using 0.4 N NaOH. The membranes were then reprobed with primary antibodies against total proteins. Western blot was performed as described above. The primary and secondary antibodies included phospho-ERK1/2(Thr202/Tyr204) rabbit antibody (#4370S, Cell Signaling Technology), ERK1/2 mouse antibody (#4696S, Cell Signaling Technology), phospho-p38 (Thr180/Tyr182) mouse antibody (#9216S, Cell Signaling Technology), p38 rabbit antibody (#9212S, Cell Signaling Technology), phospho-JNK (Thr183/Tyr185) mouse antibody (#9255S, Cell Signaling Technology), JNK rabbit antibody (#9252S, Cell Signaling Technology), β-actin mouse antibody (A5441, Sigma-Aldrich), anti-mouse IgG HRP antibody (#7076S, Cell Signaling Technology), and anti-rabbit IgG HRP antibody (#7074S, Cell Signaling Technology).
3) Quantification of protein expression pattern
Densitometric analysis was performed on protein bands using ImageJ software (https://imagej.nih.gov), and the relative values were determined.
4. [Ca2+]i concentration measurement
The Fura-2 AM calcium flux assay kit (ab176766, Abcam) was used to measure the [Ca2+]i concentration. The experimental procedure and reagents, including Fura-2 AM, were performed and used according to the manufacturer's protocol. Before the assay, a 96-well plate (black well/clear bottom, Corning) was treated with 0.1% gelatin solution (#ES-006-B, Millipore) for 1 hour. After removing the gelatin solution, 2×104 cells were seeded per well in duplicate. After 2 days, cells were transfected with 0.2 μg of either WT or mutant CASR expression vector mixed with 2.5× lipofectamine, and the existing medium was replaced with complete medium (DMEM, 10% FBS, 1×PS). Before calcium measurement, the cells were incubated for 24 hours after replacing the medium with calcium-free DMEM, then were transfected. At 48-hour posttransfection, 100 μL of solution containing Fura-2 AM was added, and the cells were treated for 1 hour at 37℃. A microplate reader (Synergy H4 Hybrid Multi-Mode Microplate Reader, BioTek, Winooski, VT, USA) was used for baseline measurements for Ex/Em=340/510 nm and Ex/Em=380/510 nm. After baseline measurements, the cells were loaded with different concentrations of calcium solution, and a kinetic assay was performed until the graph of the measured wavelength achieved a steady-state peak on the microplate reader. The reaction responses were approximately 60–90 seconds after calcium stimulation, and the maximum response was 120–180 seconds. All reactions were observed in kinetic reaction mode for 15 minutes. The 340 nm/380 nm ratio for all measurements was calculated, and [Ca2+]i values were compared on a graph.
Results
1. Expression of CASR-WT and CASR-Y825F in HEK293 cells
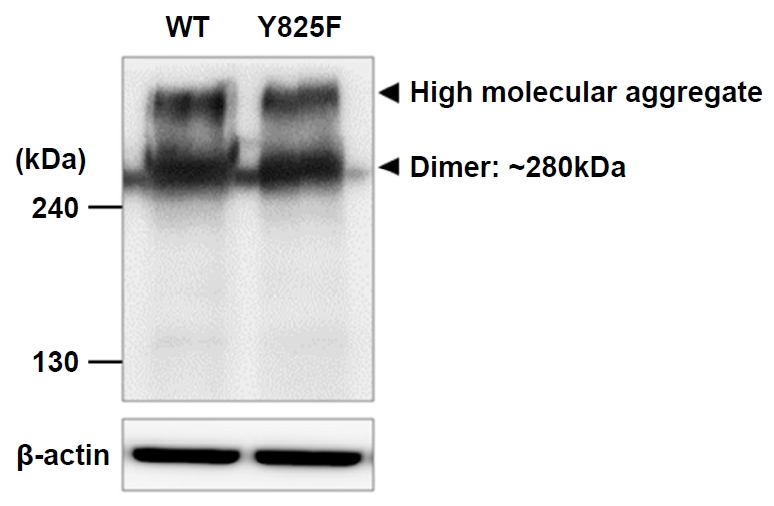
The molecular weight of the CASR monomer is approximately 130 kDa, and the expression of CASR as a dimer on transformed HEK293 cell surfaces has been reported [12]. To confirm whether there is a structural difference between CASR-Y825F and CASR-WT proteins, we generated expression vectors for CASR-WT and CASR-Y825F and transfected the plasmids into HEK293 cells. Western blotting was then performed using CASR antibodies. Consistent with the previous study that CASR-WT is generally expressed as a dimer, we found that CASR-WT existed as a higher-order oligomer (high molecular aggregate) on the HEK293 cell surface. CASR-Y825F showed an identical pattern as the CASR-WT, which confirmed that there was no difference in molecular mass (Fig. 1).
2. MAPK signaling activation in HEK293 cells expressing CASR-WT or CASR-Y825F
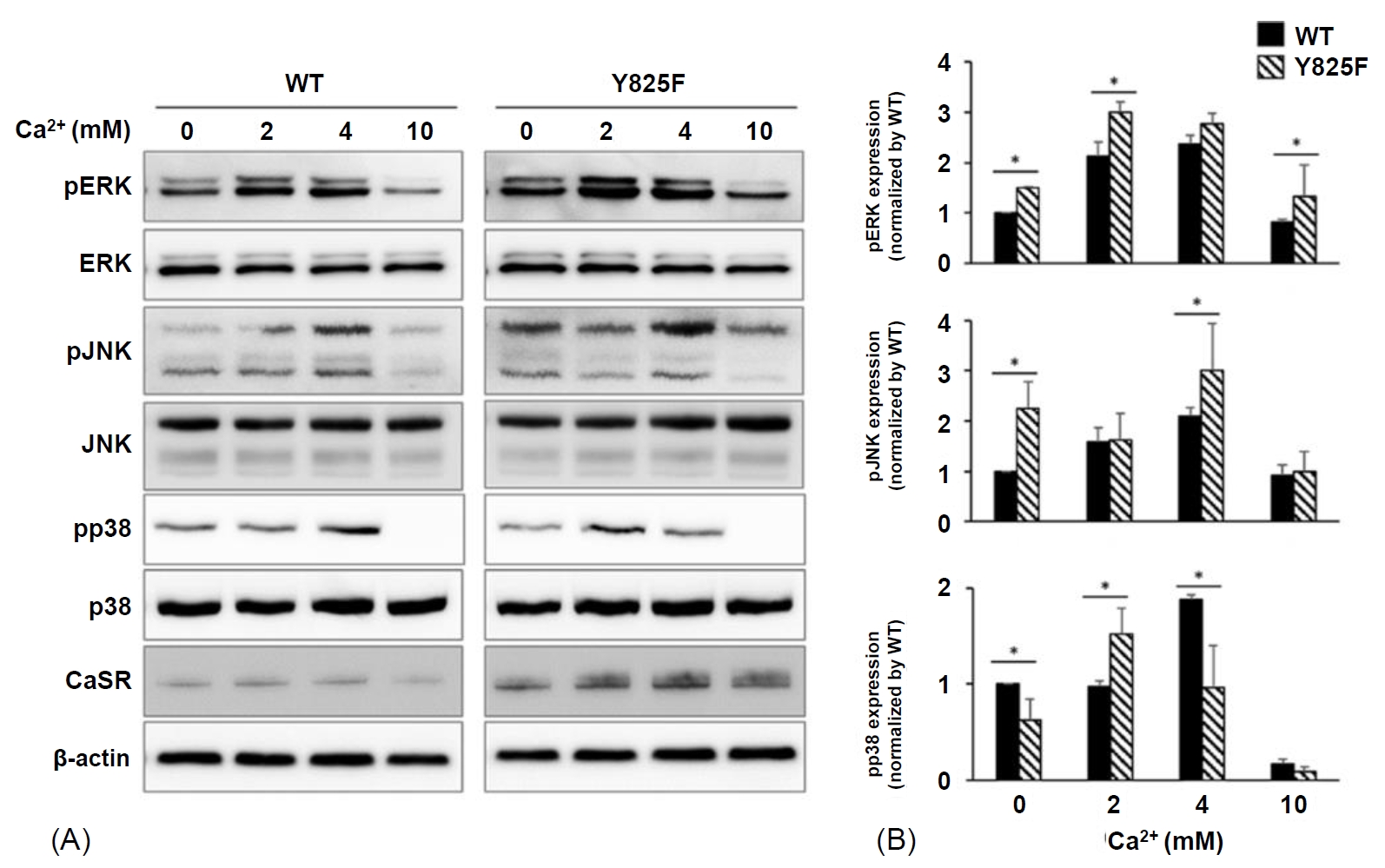
Western blot analysis was performed to determine MAPK signaling activation by CASR-Y825F and CASR-WT in HEK293 cells. CASR-Y825F and CASR-W T plasmids were transfected into HEK293 cells treated with various concentrations of [Ca2+]e (0, 2, 4, 10 mM). The degree of activation of MAPK signaling was determined using antibodies against phosphor-ERK (Thr202/Tyr204), phosphor-JNK (Thr183/Tyr185), and phosphor-p38 (Thr180/Tyr182), which detect the phosphorylation of ERK, JNK, and p38, respectively. With no stimulation of calcium, ERK and JNK phosphorylation were higher in HEK293 cells expressing CASR-Y825F than those expressing CASR-WT (Fig. 2A, B). At 2 mM calcium, MAPK activation was significantly increased in HEK293 cells expressing CASR-Y825F compared with cells expressing CASR-WT (P<0.05) (Fig. 2B). At 4 mM calcium, the degree of ERK and JNK phosphorylation was still higher in HEK293 cells expressing CASR-Y825F, unlike p38 phosphorylation (Fig. 2B). However, at 10 mM calcium, the phosphorylation of MAPK did not show a normal pattern due to cell damage. These results confirmed that activation of MAPK signaling by CASR-WT and CASR-Y825F varied depending on the concentration of [Ca2+]e. The MAPK signaling activity was higher in HEK293 cells expressing CASR-Y825F compared with cells expressing CASR-WT, indicating that MAPK was hyperactive in response to CASR-Y825F.
3. Changes in [Ca2+]i concentration in HEK293 cells expressing CASR-WT or CASR-Y825F in response to [Ca2+]e stimulation
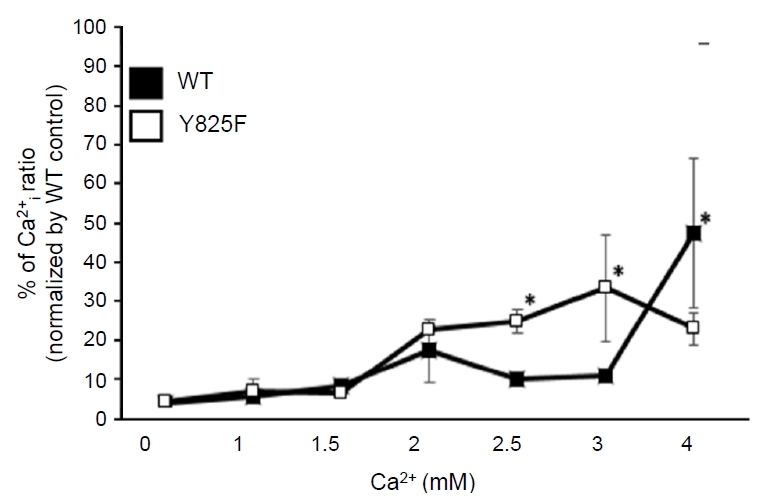
The [Ca2+]i of CASR-WT and CASR-Y825F in HEK293 cells was determined at 340 nm/380 nm wavelength ratios using Fura-2 AM. The [Ca2+]i level of HEK293 cells transfected with CASR-WT plasmid was set as the baseline value and compared with the [Ca2+]i level of the CASR-Y825F-expressing cells. The 340 nm/380 nm wavelength ratios were higher in HEK293 cells transfected with CASR-Y825F plasmid treated with 2 mM, 2.5 mM, and 3 mM [Ca2+]e. Specifically, when cells expressing CASR-WT were treated with 2.5 mM and 3 mM [Ca2+]e solution, the wavelength ratios were 1.04 and 1.40, respectively, whereas the wavelength ratios of cells expressing CASR-Y825F were 2.6 and 3.5, respectively (P<0.05). When the cells were treated with 4 mM [Ca2+]e solution, the [Ca2+]i was higher in HEK293 cells expressing CASR-WT than those expressing CASR-Y825F (Fig. 3). Together, these findings indicate that the [Ca2+]i level of HEK293 cells expressing CASR-Y825F was higher at medium concentrations of [Ca2+]e, between 2 mM and 4 mM, than cells expressing CASR-WT.
Discussion
Here we performed in vitro functional analyses using HEK293 cells ectopically expressing the CASR-Y285F mutant (CASR-c.2474A>T; p.Tyr825Phe) to determine the pathogenicity of this novel variant. Changes in [Ca2+]i and [Ca2+]e concentrations and increased MAPK signaling were experimentally examined in cells expressing the variant compared with those expressing wild-type protein.
In general, the causes of hypocalcemia occurring within 72 hours after birth include preterm birth, fetal growth retardation, maternal diabetes, neonatal asphyxia, excessive phosphate consumption, and hypomagnesemia-induced hypoparathyroidism [14,15]. The CASR variant (p.Tyr825Phe) found in the previous case report was a novel variant in ADHH [12]. The variant Y825F that the author had reported in 2018 was a novel variant of the TM domain region that had been previously determined as pathogenic and producing identical clinical patterns (hypocalcemia, hypomagnesemia, and hypercalciuria accompanied by hypoparathyroidism). The novel variant Y825F affects a region very close to previously reported pathogenic variants, A824P and T828N [8].
Increased MAPK activation by activating CASR mutation has been demonstrated in many previous studies [16-19]. The P136L mutation, reported by Pearce et al., caused increased sensitivity to [Ca2+]e stimulation [20]. Bräuner-Osborne et al. [21] published a study demonstrating that [Ca2+]e and the CASR complex activate intracellular signaling pathways, including the MAPK pathway. In human embryonic kidney cells that constitutively express CASR, ERK1 and ERK2 become activated in the presence of low concentrations of [Ca2+]e (0.5 mM CaCl2), and CASR-dependent ERK1 and ERK2 are quickly activated and then become desensitized after 10 min at a [Ca2+] e concentration of 4 mM [22-25]. Consistent with the finding in our study that the CASR response was detected at 4 mM calcium, the [Ca2+]i levels of CASR-WT cells increased at a [Ca2+]e concentration of 4 mM, and cells expressing CASR-Y825F also showed higher [Ca2+]i stimulation at low concentrations of [Ca2+]e compared with CASR-WT cells. Additionally, MAPK activation assays revealed increased phosphorylation of ERK, JNK, and p38 at low concentrations of [Ca2+]e in cells expressing CASR-Y825F compared with those expressing CASR-WT. Together, these findings revealed that cells expressing CASR-Y825F responded quickly and sensitively to low concentrations of [Ca2+]e stimulation. Western blot analysis of cells expressing CASR-WT showed that phospho-p38 (pERK), phosphoJNK (pJNK), and phospho-p38 (pp38) were activated at low concentrations of [Ca2+]e. Bai et al. [26] reported that cells expressing WT CASR showed an increase in calcium sensitivity at 4 mM compared with 3.3 mM for mutant CASR. Our study also showed the [Ca2+]i levels of CASR-WT cells increased at an [Ca2+]e concentration of 4 mM, while that of CASR-Y825F cells increased at an [Ca2+]e concentration of 2~3 mM (Fig. 3). This demonstrates that CASR-Y825F increased calcium sensitivity at a lower [Ca2+]e concentration than CASR-WT.
Leach et al. [27] reported that inactivating mutations in CASR result in equal signaling via the Ca2+ and MAPK pathways or predominantly via MAPK pathways, while many activating mutations of CASR couple more strongly to Ca2+. According to the study by Zhao et al. [28], the A843E mutation in TM7 functionally characterized a CASR mutation and caused constitutive activation rather than enhanced calcium sensitivity. This result differed from the previously reported CASR of TM that had an enhanced sensitivity depending on the calcium concentration. In this study, cells expressing CASR-Y825F, as a variant in TM7, did not show elevated MAPK activity and [Ca2+]i concentration at above 4 mM [Ca2+]e concentration compared with cells expressing CASR-WT; however, elevated reaction was observed at concentrations below 4 mM [Ca2+] e. We speculate that one possibility for the absence of western blot bands at 10 mM was because of low cell stability at a high [Ca2+]e concentration. However, there could also be causing a constitutive activation that does not depend on a calcium concentration at above 4 mM [Ca2+]e concentration. The activation of different MAPK pathways (pERK, pJNK, and pp38) in response to variable [Ca2+]e concentrations with activated CASR mutants has not been well studied. Our findings showed that the degree of phosphorylation of ERK and JNK was still higher in HEK293 cells expressing CASR-Y825F compared with those expressing CASR-WT, unlike the degree of activation of p38, at a [Ca2+]e concentration of 4 mM. Therefore, the increased activity of MAPK and increased [Ca2+]i at the low (< 4 mM) [Ca2+]e concentration may be driven by the activation of pERK and pJNK.
One limitation of this study is that we did not confirm MAPK signal activation at calcium ion concentrations between 4 mM to 10 mM; this study was designed to examine MAPK activation of CASR-Y825F-expressing cells at low [Ca2+]e concentrations below 4 mM. Cells expressing CASR-Y825F or CASR-WT both indicated a low level of MAPK activation at 10 mM. We observed that when the cells were treated with 10 mM of calcium, the cells on the bottom were dripped and the pellet volumes were reduced during sampling, suggesting that the presence of cell damage because of the high concentration calcium treatment. To clarify the response of CASR variants containing mutations in the TM domains like Y825F, further studies on changes in intracellular calcium and protein structure studies are required.
In conclusion, this study revealed that CASR-Y825F expressed in HEK293 cells elevated MAPK signaling (pERK, pJNK, and pp38) and increased [Ca2+]i to induce hypocalcemia. Further research is needed to study whether changes in protein structure lead to differences in MAPK activation in pediatric patients with the activating variant of CASR.