Introduction
Amenorrhea is defined by the transient or permanent absence of menstrual flow. Amenorrhea can be divided into primary and secondary presentations. Primary amenorrhea is diagnosed when there is no menarche by 13 years of age in those without development of secondary sexual characteristics, or by 15 years of age in those with secondary sexual characteristics [1]. Secondary amenorrhea is defined as discontinuation of previously regular menses for 3 months or previously irregular menses for 6 months [2]. The prevalence of primary amenorrhea is less than 0.1%, and secondary amenorrhea is more common with an incidence of about 4.0% [3,4].
Primary amenorrhea can result from many different underlying conditions. According to the etiology, amenorrhea can be categorized as: outflow tract abnormalities, primary ovarian insufficiency, hypothalamic or pituitary disorders, other endocrine gland disorders such as thyroid or adrenal gland disorders, and other causes (Table 1) [5]. In a previous study, the most common cause of primary amenorrhea was reported as gonadal dysgenesis, including Turner syndrome (43%), followed by Müllerian agenesis (15%) and constitutional delay of puberty (14%) [6].
Regarding various etiologies, it is important to evaluate amenorrhea carefully not to miss the underlying pathology. In this article, differential diagnoses and management of primary amenorrhea are reviewed with special emphasis on congenital sex hormonal disorders.
Evalution
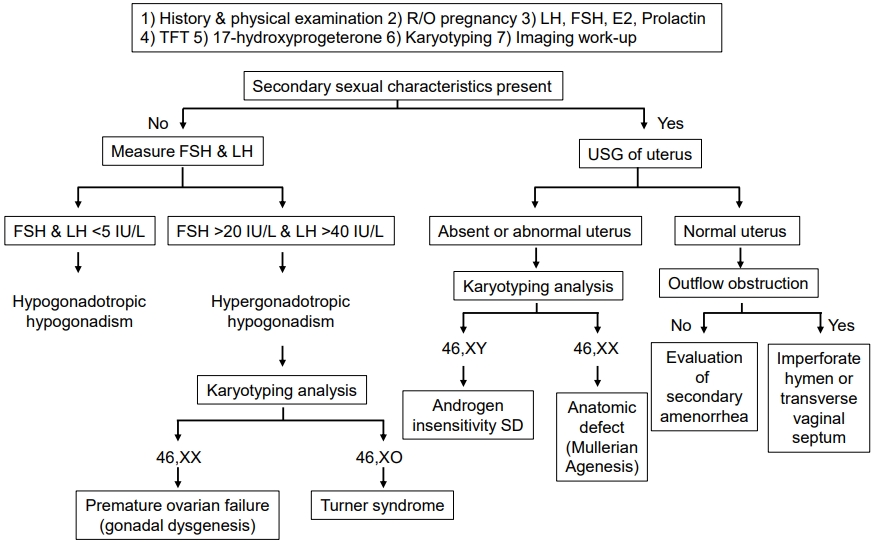
Traditionally, clinicians conduct history taking, physical examinations, endocrine tests, and radiological imaging to evaluate primary amenorrhea.
1. History
History taking about the possibility of pregnancy, breastfeeding history, eating and exercise habits, psychosocial stressors (e.g., perfectionist behaviors), changes in body weight, fractures, medication, chronic illness, and onset age of second sexual characteristics development should be performed (Table 2, Fig. 1) [5]. History of galactorrhea suggests excess prolactin which can be caused by hypothalamic or pituitary diseases or by drugs, such as metoclopramide and antipsychotic drugs. Headaches or visual field defects also can indicate underlying hypothalamic or pituitary disease [7]. A family history should be investigated including the age of menarche of the patient’s mother and siblings.
2. Physical examination
Clinicians should investigate changes in growth parameters such as height, weight, and body mass index (Table 2). Breast development indicates that an adequate amount of circulating estrogen exists [5].
Physical examination (or ultrasonography if needed) should be performed to investigate the presence of the uterus (Fig. 1). In addition, the vagina and cervix should be examined for anatomic abnormalities. A vaginal exam can determine the presence of outflow tract obstruction or Müllerian agenesis [8]. Acne or hirsutism suggests polycystic ovary syndrome (PCOS), while virilization can imply a more severe androgen excess due to an androgen-secreting ovarian or adrenal tumor or 5-alpha-reductase deficiency (5α-RD2) [9]. Dysmorphism such as the presence of a webbed neck or shield chest may suggest Turner syndrome.
3. Laboratory and other testing
A pregnancy test is recommended as a first step in evaluating any girls with primary amenorrhea [10]. Serum levels of follicle stimulating hormone (FSH), luteinizing hormone, prolactin, and thyroid-stimulating hormone should be measured to rule out endocrine causes of amenorrhea (Table 3) [5]. If there are symptoms and signs of hyperandrogenism, serum free and total testosterone, and dehydroepiandrosterone sulfate levels should be measured. A hormonal challenge can be used to determine the presence of functioning anatomy and adequate endometrial estrogen exposure [5]. If a central lesion is suspected, magnetic resonance imaging (MRI) of the brain should be considered. Furthermore, pelvic ultrasonography or MRI can identify structural anomalies of the outflow tract [10]. In patients with short stature and primary amenorrhea, karyotyping should be considered to rule out a syndromic disorder such as Turner syndrome [8]. Moreover, 46, XY disorders of sexual development (DSD) can also present with primary amenorrhea with normal external female genitalia. Actually, the genetic causes of DSD are highly heterogeneous thereby posing a diagnostic challenge. Baxter et al. [11] reported whole exome sequencing followed by analysis of selected DSD genes as having a diagnostic yield of 22.5% in patients with 46,XY DSD. A more recent study revealed a relatively higher yield by a targeted panel sequencing approach, in which a genetic diagnosis was made in 38.1% [12]. Considering the diagnostic potential of a high-throughput approach, next-generation sequencing (NGS) panels for DSD could be used in practice to uncover the underlying genetic causes of primary amenorrhea.
Anomalies of the outflow tract
1. Imperforate hymen
An imperforate hymen usually presents as a bluish bulging mass due to hematocolpos at the entrance to the vagina. The treatment is simple resection to allow for drainage of menstrual blood, and then the hymen is excised. After treatment, patients can have normal menstrual function.
2. Transverse vaginal septum
A transverse vaginal septum is caused by persistence of the vaginal plate after it meets the Müllerian tract [13]. Septa can lie anywhere along the length of the vaginal cavity. The most common location is high in the vaginal cavity [14]. It is a rare anomaly, found in 1:2100 to 1:72,000 females [15]. The physical exam reveals a shortened blind vaginal pouch. Symptoms include dysmenorrhea and/or dyspareunia. The treatment is surgical resection.
3. Müllerian agenesis
Müllerian agenesis, also known as vaginal agenesis or Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome, is defined by congenital absence of the vagina. It is most commonly associated with uterine agenesis, but, 7% to 10% of patients have an obstructed or rudimentary uterus [15].
Patients will typically have no complaint other than amenorrhea. If the patient complains of cyclic pain, MRI is useful in detecting endometrium in uterine remnants. Remaining active endometrial tissue may require surgical removal. Patients have normal female external genitalia, ovaries, and hormonal patterns. Patients can also have renal and skeletal anomalies. MRKH syndrome accounts for 20% of the causes of primary amenorrhea [16] in Koreans.
Gonadal dysgenesis
Gonadal dysgenesis refers to incomplete or defective formation of the gonads, which is the most common cause of primary amenorrhea (30%–40%) [13]. These women have decreased estradiol and an elevated FSH level. There are many types of gonadal dysgenesis: pure, partial, and mixed. Pure gonadal dysgenesis patients have streak gonads with karyotypes such as 45,XO (Turner syndrome), 46,XX, or 46,XY (Swyer syndrome). Patients with partial gonadal dysgenesis have bilateral dysgenic gonads, and mixed gonadal dysgenesis present with 1 streak and 1 dysgenic gonad.
1. Turner syndrome
About two-thirds of cases of gonadal dysgenesis are caused by Turner syndrome [17]. In Turner syndrome, while the ovarian germ cells undergo normal migration and mitosis, normal meiosis does not occur, leading to rapid loss of oocytes. About 70%–80% have no spontaneous pubertal development and 90% experience primary amenorrhea [18]. Estrogen and cyclic progesterone replacement is required for induction of pubertal development from about 11 years of age. Unopposed low dose estrogen should be started first, as progesterone will cause misshapen tubular breasts. Heterologous in vitro fertilization—embryo transfer, using oocyte donation, is the most used reproductive technique and it represents the only way to become pregnant. At the age of 13–14 years, girls with Turner syndrome should be counselled about fertility options and, in those with mosaic karyotype and spontaneous puberty in the absence of any elevation in FSH or reduction in AMH, the discussion should include cryopreservation [19,20]. Given the concerns with ovarian cryopreservation in Turner syndrome, an algorithmic approach to decision-making for fertility preservation in females with Turner syndrome was developed [21].
2. 46,XX gonadal dysgenesis
46,XX gonadal dysgenesis is caused by defects in autosomal genes. Patients with 46,XX gonadal dysgenesis present with amenorrhea and absent secondary sex characteristics, but otherwise have a normal phenotype [13]. Patients have a normal 46,XX karyotype and streak gonads [22]. There are syndromic forms accompanying sensorineural hearing loss (Perrault syndrome). hormone replacement therapy (HRT) with estrogen and cyclic progesterone is required.
3. 46,XY gonadal dysgenesis (Swyer syndrome)
46,XY gonadal dysgenesis is also called Swyer syndrome. About 10% to 20% of these cases are caused by SRY gene mutation [13]. Patients have streak gonads or nonfunctional testicular tissue.
The gonads produce neither AMH nor androgens. As a result, they have normal female internal and external genitalia. They are phenotypically female, and present with primary amenorrhea and delayed secondary sex characteristics. Gonadectomy is required due to a high risk of malignant transformation. Patients also receive HRT for induction of puberty. Pregnancy is possible with appropriate HRT and assisted reproductive technology [23].
Central anomalies
1. Pituitary disorders
Hyperprolactinemia or decreased gonadotropin due to disturbance of pituitary function can cause amenorrhea.
1) Hyperprolactinemia
Hyperprolactinemia can be caused by multiple causes (prolactinoma, diseases involving the hypothalamus-pituitary, medications, systemic illnesses, etc.) and is a common cause of amenorrhea.
Hyperprolactinemia accounts for 4% of the causes of primary amenorrhea in Koreans [16]. Patients with a prolactinoma may have galactorrhea or ocular headaches [13]. Patients with a macroadenoma may also present with symptoms of pituitary stalk compression such as headaches, visual disturbances, poor growth, and diabetes insipidus [22]. Dopamine agonists are used for the treatment of hyperprolactinemia. They decrease prolactin, resume menses, increase fertility, and increase absorption of calcium to bones. Combined oral contraceptives can be used to restore menstrual regularity.
2) Empty sella syndrome
Empty sella syndrome is defined by the shrinkage or flattening of the pituitary gland. This disorder is due to congenital defects in the sellar diaphragm in which the sella turcica is filled with cerebrospinal fluid instead of the normal pituitary gland. This condition can cause pituitary stalk compression, disrupting the hypothalamic-pituitary-ovarian axis [13].
2. Hypothalamic disorders
Hypothalamic disorders can cause hypogonadotropic hypogonadism. Etiologies include craniopharyngioma, cranial irradiation, functional hypothalamic amenorrhea, Kallmann syndrome, etc.
1) Craniopharyngiomas
Craniopharyngiomas account for 3% of intracranial neoplasms and 1% of primary amenorrhea [24].
Most present at ages of 6 to 14 years with symptoms such as headaches, visual disturbances, delayed development, poor growth, and diabetes insipidus [22]. Amenorrhea is caused by compression of the hypothalamus and pituitary. Treatment is surgical resection, but recurrence is common. The addition of radiation and microsurgical techniques can decrease the recurrence rate [24].
2) Functional hypothalamic amenorrhea
Functional hypothalamic amenorrhea is caused by disorders that inhibit pulsatile release of GnRH, such as eating disorders, exercise, malnutrition, and stress. They are usually causes of secondary amenorrhea but can cause primary amenorrhea. Amenorrhea from these disorders, to some extent, is explained by a stress-induced increase in cortisol levels due to activation of corticotrophin-releasing hormone [13].
3) Kallmann syndrome
Kallmann syndrome is an X-linked disorder, which is characterized by delayed puberty and anosmia. Kallmann syndrome results from mutations that cause a defect in migration of the GnRH neurons and the olfactory neurons [22]. Treatment includes sex hormone replacement with estrogen and progesterone. Gonadotropin or pulsatile GnRH therapy can also be used to induce fertility [25].
Receptor abnormalities and enzyme deficiencies
1. Androgen insensitivity syndrome
Androgen insensitivity syndrome (AIS) is caused by defective function of the androgen receptor. An inactivating mutation of the gene encoding for the androgen receptor leads to endorgan insensitivity to androgens. Androgen resistance that results in complete AIS is characterized by XY sex reversal with a normal female phenotype. With residual androgen receptor activity, partial AIS results in a variable phenotype. The majority of 46,XY DSD cases are due to AIS with elevated testosterone levels. Anti-Müllerian hormone (AMH) is normally secreted, resulting in a short blind vagina. There is breast development due to peripheral conversion of androgens to estrogen [13]. Amenorrhea is the most clinically observed sign of AIS. Kwon et al. [16] reported AIS in 8.3% among 132 patients with primary amenorrhea in Korea. Removal of the gonads owing to a 5% to10% incidence of malignancy is required [13]. However, gonadectomy should be recommended after completion of puberty because tumor incidence is very rare in patients under 20 years of age [26]. After gonadectomy, hormone replacement therapy is required in order to maintain secondary sexual characteristics, bone and cardiovascular health and to promote general wellbeing and sexual function [27].
2. 5-Alpha-reductase deficiency
5-Alpha-reductase deficiency can result in primary amenorrhea in a 46, XY subject. At birth, these patients may have ambiguous genitalia due to an inability to convert testosterone to its more potent metabolite dihydrotestosterone (DHT). Two-thirds of patients with 5α-RD2 who were initially assigned female gender and all who were assigned male gender, live as males [28]. During puberty, they can exhibit virilization due to the normal peripubertal increase in testosterone secretion without breast development [29]. However, these individuals do not undergo enlargement of the male external genitalia which is DHT-dependent [30]. Management decisions are based on multiple factors including reproductive anatomy, DSD etiology, parental/cultural factors, and most importantly, outcome [31]. The size of the penis and its potential to develop at puberty into a sexually functional penis are among the most important concerns when one is considering assignment of male sex [32]. If the patient is to be raised as a male, surgical correction should be performed. Standard surgical repair operations such as chordee correction, orchidopexia, and urethral reconstruction are done between 6 and 18 months of age, usually in one stage as an outpatient procedure [28]. Prior to surgery, treatment with androgens is required to increase phallic length and promote hypospadia repair. Meanwhile, if the child is raised as a female, surgical correction of the external genitalia should be considered and gonadal tissue should be removed early to prevent masculinization before puberty [33]. Feminizing genital surgery includes reconstruction of the external genitalia and providing an adequate vaginal opening into the perineum, with early separation of the vagina and urethra [28,34]. During the teenage years, vaginoplasty should be considered [28]. Cyclic hormonal therapy at puberty for the development of secondary sexual characteristics is required.
3. FSH receptor mutation
Inactivating mutations of the FSH receptor (FSHR) can cause amenorrhea due to ovarian failure [35]. In individuals with FSH resistance, follicular maturation is impaired, although most patients show small (3–5 mm) follicles by transvaginal ultrasound and follicular arrest at the small antral stage [36,37]. As a result, inactivating mutations of FSHR cause hypergonadotropic hypoestrogenic amenorrhea with elevated FSH and low estrogen levels. In patients with a FSH receptor mutation, AMH is secreted by small growing follicles, resulting in detectable values which are likely varying with the mutation severity and follicular arrest stage [38]. Because FSHR mutations are detected in <1% of patients with primary ovarian failure [39], a candidate gene approach is far less efficient than whole exome sequencing (WES) for uncovering the etiology of primary amenorrhea. WES-assisted diagnosis allows for treatments aimed at the underlying molecular etiology of disease. Analysis of genotype–phenotype association and in vitro functional characterization of inactivating mutations of FSHR have led to important information about the contribution of the mutated residues and the corresponding region in hormone receptor interaction and signal transduction [40]. Therefore, research on pharmacological and assisted reproductive treatments aimed at the disrupted FSHR is necessary to provide patients with FSH resistance personalized medicine.
4. Polycystic ovary syndrome
PCOS is characterized by ovulatory dysfunction, biochemical or clinical androgen access, and polycystic ovaries [8]. FSHR is known to play a role in the pathophysiology of the disease. Polymorphisms of FSHR are known to alter the response to exogenous FSH or the risk of having PCOS [41]. In addition, genome wide association studies have found that variants in the FSH-β gene and the LH receptor gene (LHCGR) were also found by genome wide association study [42].
The American Society of Reproductive Medicine Practice Committee reported that 1 of the 4 most common cause of amenorrhea was PCOS [43]. Prescribing combined oral contraceptives is the first-line therapy for menstrual abnormalities, hirsutism, and acne. Oral contraceptives can also provide protection from endometrial cancer caused by unopposed estrogen secretion [44,45]. Metformin is recommended for patients with impaired glucose tolerance and those in whom lifestyle modifications are unsuccessful, or for those with contraindications to applicable contraceptives [44]. For patients with PCOS and infertility, letrozole is the treatment of choice, because higher rates of ovulation, pregnancy, and live birth were reported with letrozole than clomiphene [44].
5. Nonclassic congenital adrenal hyperplasia
Nonclassic congenital adrenal hyperplasia (NCAH) is a common cause of hyperandrogenic amenorrhea. NCAH due to P450c21 (21-hydroxylase) deficiency is a common autosomal recessive disorder due to mutations in the CYP21A2 gene. The clinical consequences of NCAH expand from infancy, i.e., accelerated growth, to adolescence and adulthood, i.e., premature pubarche, cutaneous symptoms, hirsutism, amenorrhea, chronic anovulation, and infertility [46]. In a study of 220 adolescents, menstrual irregularities (56%) or even primary amenorrhea (9%) were the presenting sign of NCAH [47]. Despite the adrenal and ovarian androgen excess, the majority of women with NCAH will conceive spontaneously [48]. Between 10% and 30% of NCAH women of reproductive age complain of infertility [49]. Persistent elevated progestogen concentrations owing to excess circulating levels of progesterone and 17hydroxyprogesterone (17-OHP) of adrenal origin may also result in an unfavorable cervical mucus and a persistent decidualized or hypo- or atrophic endometrium [50].
6. 17-hydroxylase/17,20-lyase deficiency
This is a rare form of congenital adrenal hyperplasia, characterized by hypertension and sexual infantilism and caused by loss-of-function mutations in CYP17A1. The CYP17A1 enzyme catalyzes both steroid 17-hydroxylase and 17,20-lyase activities. 17-hydroxylase/17,20-lyase deficiency are forms of CAH that impair both adrenal and gonadal function [51]. Isolated 17,20-lyase deficiency does not significantly lead to adrenal hyperplasia and is more a form of gonadal insufficiency. Nearly 100 disease-causing mutations in 17-hydroxylase/17,20-lyase deficiency have been found. Primary amenorrhea is typical in patients with complete 17-hydroxylase/17,20-lyase deficiency. Diagnosis of 17-hydroxylase/17,20-lyase deficiency is made based on the presence of hypertension, elevated DOC (>1 ng/mL [>3 nmol/L]), elevated corticosterone (>40 ng/mL [>116 nmol/L]) with low cortisol (<5 μg/dL [<138 nmol/L]), androgens and estrogens, and suppressed renin [52,53]. Treatments of 17-hydroxylase/17,20-lyase deficiency include mineralocorticoid receptor antagonists (spironolactone), antihypertensive drugs (amiloride), and glucocorticoid [53]. HRT is initiated during adolescence or upon diagnosis if an adult. Surgery of the external genitalia is rarely necessary.
Conclusions
Here we provided a summary of the assessment and management of primary amenorrhea with special emphasis on congenital sex hormonal disorders. Despite traditional approaches such as careful history taking, physical exam, laboratory testing including chromosome and/or single gene analysis, and imaging work-up to evaluate primary amenorrhea, the molecular basis and pathophysiology remains unknown for some rare causes of primary amenorrhea. These rare causes may call for NGS-based methods allowing for a broad search for variants in patients with puzzling phenotypes. Treatment of primary amenorrhea may vary considerably, and rely on the patient and the specific diagnosis.