Introduction
Mayer-Rokitansky-Küster-Hauser syndrome (MRKH; Online Mendelian Inheritance in Man (OMIN) #277000) is characterized by developmental failure of the Müllerian duct and is often found in combination with urological and skeletal anomalies [1,2]. Müllerian duct aplasia-renal aplasia-cervicothoracic somite dysplasia (MURCS) association (OMIN #601076), which could be classified as a subtype of MRKH, is a unique development disorder with four common types of malformations, including uterine aplasia or hypoplasia, renal ectopy or agenesis, vertebral anomalies and short stature (adult height less than 152 cm) [1,3]. MURCS association was first defined by Duncan et al. in 1979 [3] as congenital supravaginal and uterus, renal and cervicothoracic abnormalities. Other organ deformities were subsequently reported, such as external ear anomalies, deafness, facial asymmetry, and cardiac defects [4].
In affected patients, the classical presentations of MUCRS, as demonstrated in cases with MRKH [5], are primary amenorrhea with normally-developed secondary sexual characteristics and normal karyotype. Therefore, the majority of the MURCS patients have been diagnosed with primary amenorrhea since late-adolescence. To date, few cases of MURCS association have been diagnosed during childhood and long-term outcomes are not well reported. We report a case of an 8-year-old girl with MURCS association who presented with severe short stature, recurrent urinary tract infection, and multiple congenital malformations who was followed for 10 years until adulthood, and we present a review of the literature.
Case report
An 8-year-old girl with a history of renal anomaly was referred to our hospital for evaluation of other organ deformities. The patient had lived in an orphanage since birth. Family and prenatal histories were not confirmed. She had undergone open-heart surgery due to a ventricular septal defect at the age of 6 months at another hospital. The patient was frequently treated for recurrent urinary tract infections. At the age of 4 years, she underwent an operation due to a diagnosis of vesicoureteral reflux (VUR) and a single dysplastic kidney in the pelvis.
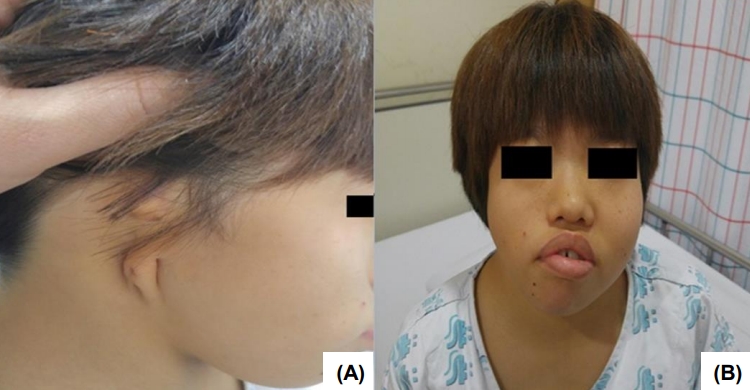
On admission, the vital signs were as follows: systolic and diastolic blood pressure 118 (95th percentile) and 65 mmHg (75th–95th percentile); pulse rate 70 times/min.; respiratory rate 20 times/min.; body temperature 36.2℃. Body weight was 13.3 kg (-5.42 standard deviation score [SDS]) and height was 91.2 cm (-7.91 SDS). Physical examination revealed facial asymmetry, a short neck, external ear aplasia on the right side (Fig. 1), and right thumb hypoplasia. She had normal female external genitalia and showed normal intellectual development. Initial blood examination results were as follows: hemoglobin, 10.7 g/dL; leukocyte, 8,020/μL; platelet count, 355×103/mm; serum total protein, 8.7 g/dL; serum albumin, 4.7 g/dL; total cholesterol, 246 mg/dL; aspartate aminotransferase, 38 IU/L; alanine aminotransferase, 21 IU/L; serum sodium, 141 mEq/L; potassium, 4.9 mEq/L; chloride-, 109 mEq/L. The blood urea nitrogen (BUN) and serum creatinine were 31.9 mg/dL and 1.40 mg/dL, respectively, and the glomerular filtration rate (by calculated Schwartz formula) was 35.8 mL/min/1.73 m2. The urinalysis showed normal findings. The karyotype was normal female as 46, XX. The serum insulin-like growth factor 1 level was within the normal range as 186.0 ng/mL (normal range, 100–446 ng/mL).
A growth hormone (GH) stimulation test was performed to evaluate her extremely short stature. In the levodopa provocative test, the GH levels showed an exaggerated increase from 3.98 ng/dL to 46.5 ng/dL compared to the GH deficiency. The spine X-ray showed a fusion of C2–3, and C4–5 vertebrae as well as a fusion of L1–2, and L3–4 vertebrae. The costal X-ray showed that the 1st and 2nd ribs on both sides were fused. The X-ray also showed scoliosis and sacrococcygeal agenesis (Fig. 2). Abdominal computed tomography revealed a single left dysplastic kidney, located in the pelvis, and uterine aplasia (Fig. 3). Evaluation of the auditory system revealed sensorineural hearing loss of the right ear as well as external ear malformation. Based on the co-occurrence of all deformities, the patient was diagnosed with MURCS association. She was estimated to have chronic kidney disease (CKD) stage 3b, and she underwent a suprapubic cystostomy with a Foley catheter insertion to maintain a residual renal function. The patient started dietary intervention and conservative management, including oral medications of sulfonate calcium, cholecalciferol, and sodium bicarbonate. She started wearing a hearing aid and received psychiatric counseling. At the age of 12, the patient's medical condition was maintained, and height was 107.9 cm (-6.04 SDS) and weight 17.5 kg (-5.47 SDS). On physical examination, she showed the beginnings of secondary sexual characteristics such as thelarche (Tanner stage B2). The serum BUN and creatinine were 34.0 mg/dL and 1.73 mg/dL, respectively, and the glomerular filtration rate (Schwartz formula) was maintained as 34.3 mL/min/1.73 m2. At the age of 18, her height was 132.6 cm (-5.78 SDS), her weight was 29.9 kg (-6.29 SDS), and blood pressure was 118 (50th–75th percentile)/75 mmHg (50th–75th percentile), and her pubertal development was complete, excluding menarche. Laboratory test results to evaluate her primary amenorrhea were as follows: LH was 16.0 mIU/mL (normal range, 0.5–7.6 mIU/mL), FSH was 2.43 mIU/mL (normal range, 1.2–13.4 mIU/mL), estradiol was 58.01 pg/mL (normal range, 54–242 pg/mL), dehydroepiandrosterone-sulfate was 80.3 μg/dL (normal range, 66–255 μg/dL), and the anti-Müllerian hormone was 3.43 ng/mL (normal range, 2.0–6.8 ng/mL). The renal functions had gradually declined; serum BUN and creatinine were 62.9 mg/dL and 5.05 mg/dL, respectively, and the glomerular filtration rate (Schwartz formula) was 14.4 mL/min/1.73 m2. She was scheduled to undergo continuous ambulatory peritoneal dialysis due to CKD stage 5 (end-stage renal disease) (Fig. 4). Since the procedure, she has been on peritoneal dialysis and receives conservative treatment as a renal replacement therapy.
Discussion
MURCS association is a rare and unusual disorder with multi-organ malformations and has not yet been reported in Korea. According to Guerrier et al. [4], defects of the reproductive organs are found in most cases of MURCS, while renal anomalies, including unilateral renal agenesis or ectopic or horseshoe kidney are reported in only 50% of cases. In addition, cervicothoracic vertebral anomalies are found in only 20% of cases. Other minor organ deformities are occasionally detected at a low incidence. Our patient had the 4 major anomalies of MURCS association, uterine agenesis, an ectopic single kidney with VUR, multiple vertebral anomalies, and short stature (Fig. 5), and also had other minor clinical features of a congenital heart defect, other skeletal anomalies including facial asymmetry, a short neck, and external ear malformation with deafness. In addition, our patient showed an exaggerated GH response to the GH stimulation test. To our knowledge, there has not been data reported for a GH stimulation test in MURSC association, which makes it difficult to establish a causal relationship with the disease. In this case, the rarity and clinical significance of these syndromes are highlighted (Table 1) [6-9].
MURCS association should be differentiated from various conditions such as Klippel-Feil syndrome, VACTERL association and Goldenhar syndrome due to similar accompanying anomalies. Genital deformities are rare in VATERL association, which is characterized by vertebral defects, renal anomalies, tracheoesophageal fistula, and anal atresia [10]. Klippel-Feil syndrome is typically related to facial asymmetry and vertebral anomalies, while it is rarely associated with reproductive involvement. Goldenhar syndrome can present with similar skeletal anomalies in the face, auricle, and vertebral spectrum, compared to MURCS, however urogenital deformities are rare [4].
The pathogenesis of the association between MURCS and urogenital deformities has not been elucidated. During development, at the end of the fourth week of fetal life, the blastemas of the cervicothoracic somite, arm buds, and pronephric buds are in close proximity [3]. At this time, MURCS association may develop because of a teratogenic effect, which would affect the relationship between these blastemas.
Although most cases are sporadic, some cases are reported to have a family history with autosomal dominant inheritance but incomplete penetrance. A recent genetic study on the disorder focused on various relevant genes, such as the WNT and HOX genes. The WNT genes, which encode the glycoprotein that controls the growth and differentiation of cells in the embryo stage, were expressed when female genitalia had formed in mouse experiments and mutated in cases of various Müllerian agenesis [11]. HOX genes are associated with the development of the Müllerian duct and also the kidneys and the skeleton; thus, a defect in these genes is likely to be related to MURCS association [12]. However, to date, research has not yet defined the role of these genes in the pathogenesis of MURCS [4,13].
This report demonstrates the importance of evaluating kidney or uterine anomalies via abdominal ultrasonography in patients with extremely short stature and with multiple skeletal anomalies. Management and prognosis of MURCS case varies depending on the extent of organ involvement, especially the kidneys. In this case, the severe urological anomalies were the critical prognostic factor [14]. Therefore, to ensure the quality of life in MURCS patients, it is important to delay or prevent the progression of ESRD. In conclusion, MURCS association should be considered as a differential diagnosis when evaluating prepubertal female patients with vertebral and renal malformations.