A case of de novo 18p deletion syndrome with panhypopituitarism
Article information

Abstract
Deletion on the short arm of chromosome 18 is a rare disorder characterized by intellectual disability, growth retardation, and craniofacial malformations (such as prominent ears, microcephaly, ptosis, and a round face). The phenotypic spectrum is wide, encompassing a range of abnormalities from minor congenital malformations to holoprosencephaly. We present a case of a 2-year-old girl with ptosis, a round face, broad neck with low posterior hairline, short stature, and panhypopituitarism. She underwent ventilation tube insertion for recurrent otitis media with effusion. Brain magnetic resonance imaging showed an ectopic posterior pituitary gland and a shallow, small sella turcica with poor visualization of the pituitary stalk. Cytogenetic and chromosomal microarray analysis revealed a de novo deletion on the short arm of chromosome 18 (arr 18p11.32p11.21[136,227–15,099,116]x1). She has been treated with recombinant human growth hormone (GH) therapy since the age of 6 months after diagnosis of GH deficiency. Her growth rate has improved without any side effects from the GH treatment. This case expands the phenotypic spectrum of 18p deletion syndrome and emphasizes the positive impact of GH therapy on linear growth in this syndrome characterized by growth deficiency. Further studies are required to define the genotype-phenotype correlation according to size and loci of the deletion in 18p deletion syndrome and to predict prognosis.
Introduction
The 18p deletion syndrome (Online Mendelian Inheritance in Man [OMIM] #146390) is a rare chromosomal aberration, with only approximately 150 cases reported since 1963 [1]. The prevalence of 18p deletion syndrome is estimated at 1 in 50,000 live births, with a female to male ratio of 3:2 [1,2]. There is broad phenotypic heterogeneity depending on deletion size and specific breakpoints. The most common clinical features include short stature, facial dysmorphism (e.g., epicanthal folds, a round face, ptosis, dysplastic ears, thin lips, drooped corners of the mouth, and long philtrum), and intellectual disability [3]. Skeletal and cardiac abnormalities such as congenital cardiovascular malformations have also been reported in approximately 10% of these patients [4]. Other features, including microcephaly, holoprosencephaly, clinodactyly, dystonia, short neck, and dental anomalies, have been reported less frequently [5]. Cytogenetic analysis is essential for a definite diagnosis because other disorders may have similar symptoms [1]. Most cases (more than 75%) result from de novo deletions. The remainder are caused by de novo translocation, unbalanced familial transmission of structural rearrangements due to familial translocation, inversion, complex translocation, or direct transmission [6]. Clinical manifestations tend to be associated with the size and loci of the deletion and the involved genes. However, phenotype-genotype correlation in this syndrome is still challenging.
Since the development of microarray technologies, precise deletion breakpoints and specific deleted genes in the 18p deletion syndrome have been identified [7]. Here, we report a female with 18p deletion with dysmorphic features, short stature, and panhypopituitarism.
Case report
A Korean female aged 37 days was admitted to the neonatal intensive care unit due to decreased activity, weak crying, and apnea. She was born with a weight of 2,460 g by normal vaginal delivery at 36+3 weeks of gestational age without perinatal problems. She was the first baby of healthy nonconsanguineous Korean parents. Her height was below the third percentile, and both her weight and head circumference were in the 10th percentile. The father's height was 168 cm, and the mother’s height was 162 cm. The patient showed a round face, bilateral ptosis, broad nasal bridge, long philtrum, drooped corners of the mouth, thin lips, and large, protruding ears.
Blood glucose was 16 mg/dL in capillary blood samples obtained on admission. Six milliliters of 10% dextrose solution was immediately injected as a bolus, and she showed normoglycemia. At the time of hypoglycemia, low cortisol (0.9 μg/dL [normal value, 1.8–26.0 μg/dL]), normal adrenocortical thyrotropin hormone (ACTH) (20.9 pg/mL [normal value, 0–60 pg/mL]), low free T4 (0.61 ng/dL [normal value, 0.9–2.2 ng/dL]), normal thyroid stimulating hormone (TSH) (1.811 μIU/mL [normal value, 0.7–11.0 μIU/mL]), low insulin-like growth factor (IGF)-1 level (0.1 ng/mL [normal value, 49–642 ng/mL]), and normal insulin (3.06 μIU /mL) were noted.
ACTH stimulation test revealed secondary adrenal insufficiency (cortisol basal, 0.6 μg/dL; cortisol 30 minutes, 2.0 μg/dL; cortisol 60 minutes, 0.6 μg/dL; ACTH basal, 18.1 pg/mL; ACTH 30 minutes, 15.1 pg/mL; ACTH 60 minutes, 8.5 pg/mL). For this, once daily physiologic doses of hydrocortisone (10 mg/m2) and 20 μg of levothyroxine were started.
Abdominal ultrasonogram and echocardiogram revealed no remarkable findings. Sella magnetic resonance imaging (MRI) showed a poorly visualized anterior pituitary gland and upper stalk with an ectopic posterior pituitary gland (Fig. 1).
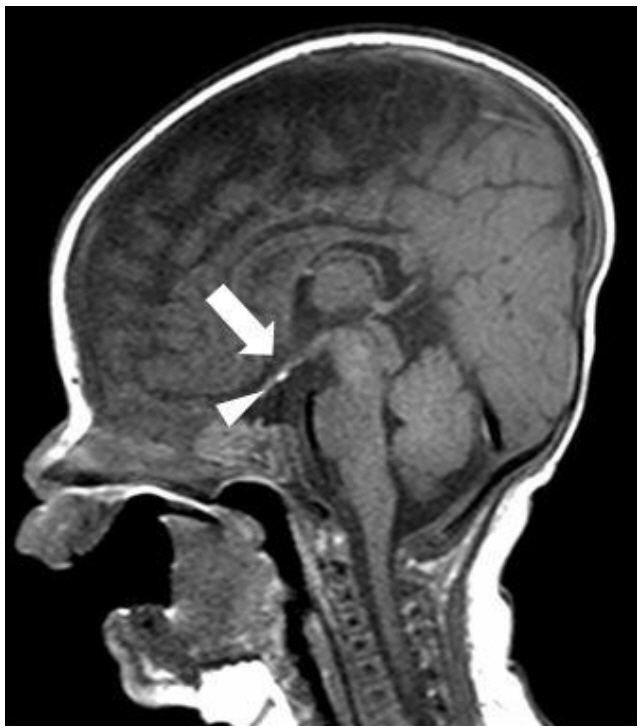
Sella magnetic resonance imaging scan of the patient at age 46 days. Unenhanced midline sagittal T1-weighted image shows poorly visualized pituitary gland in the upper stalk (arrowhead) with ectopic hypothalamic posterior gland (arrow).
Chromosomal analysis (550-band level) from peripheral blood lymphocytes revealed an unknown deletion of the short arm of chromosome 18 [46,XX,del(18)(p11.1) or der(18)t(18;?)(p11.1;?)] (Fig. 2). Chromosomal microarray analysis (CMA) was thus performed using the Affymetrix Cytoscan 750k array with reference to the human gene ver. 19 (Affymetrix, Santa Clara, CA, USA) to determine the origin of the genetic loss or gain. CMA revealed a gain in copy number on Xp22.31 of approximately 1.7 Mb and a 14.9-Mb deletion in 18p11.32p.11.21, defined as arr Xp22.31[6,446,650–8,135,568]x3 and 18p11.32p11.21[136,227–15,099,116]x1, respectively (Fig. 3A, B).
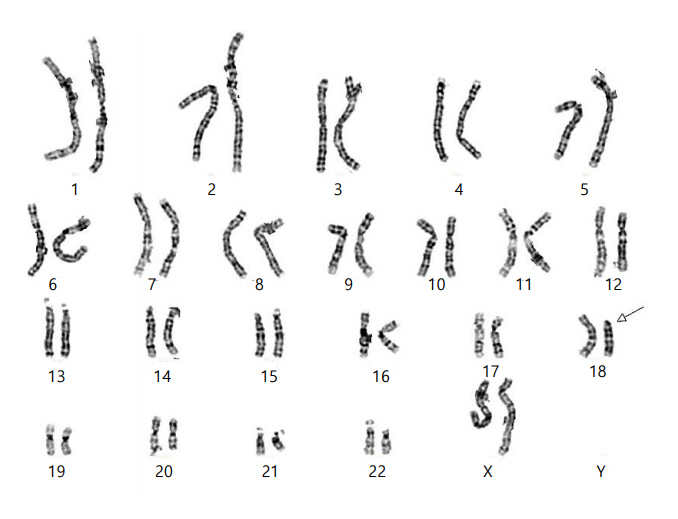
Chromosome analysis of the patient. The G-banded karyogram (550 band level) indicated deletion in the short arm of chromosome 18 (white arrow).
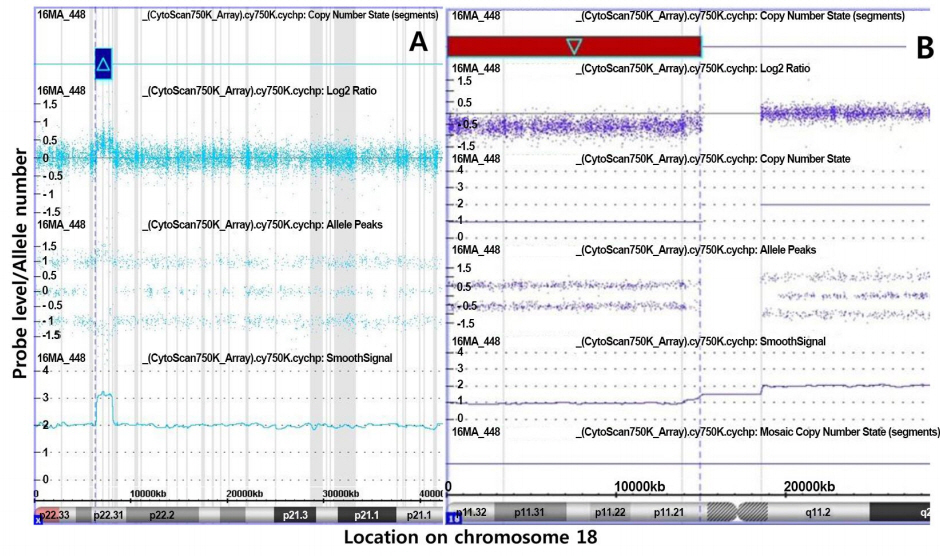
Chromosomal microarray results of the patient. (A) Array analysis indicated a 1.7-Mb duplication in Xp22.31. This copy number gain was inherited from her normal father and seems to be a benign copy number variation. (B) Array analysis indicated a 14.9-Mb (136,227–15,099,116) deletion in 18p11.32p11.21 (the deleted region is indicated by a red box with light blue outline). The X-axis represents the probe index on chromosome 18, and the Y-axis represents the signal log2 ratio of the probe.
The CMA results of the parents showed that this copy number variation (CNV) on the patient's X chromosome was derived from her father. Because the father's phenotype is normal, the CNV on the X chromosome could be benign. These findings indicate that the deletion of chromosome 18 in this patient is de novo. Therefore, the patient's karyotype was determined as follows: 46, XX, del (18) (p11.32p11.21) (arr 18p11.32p11.21[136,227–15,099,116]x1), with each metaphase having a chromosome 18 that was lacking most of the short arm from 18p11.32 to 18p11.21.
At 11 months old, the patient received bilateral ventilation tube insertion for recurrent otitis media effusion. To treat exotropia, right lateral rectus muscle recession and right medial rectus muscle resection were conducted at 12 months old. She received a perianal lipoma excision at 2 years old. Developmental milestones were within normal ranges (sitting alone at 8 months and walking at 17 months).
Because short stature and a low IGF-1 highly suggested growth hormone (GH) deficiency, recombinant human GH therapy has been administered since 6 months of age. At 3 years of age, when the patient's body weight reached 10 kg, a combined pituitary function test was performed. Basal serum concentrations of GH, TSH, and ACTH were low, and the responses of GH, TSH, and cortisol were nearly absent (hGH basal 0.14→peak 0.05 ng/mL; TSH basal 0.05→0.05 μIU/mL; cortisol basal 0.4→ peak 0.6 μg/dL). The results were consistent with panhypopituitarism. At the last follow-up at 4 years old, growth was much improved from -4.7 to -2.3 standard deviation (Supplementary Fig. 1).
Discussion
Here we describe a patient with a de novo 18p deletion presenting typical clinical features of 18p deletion syndrome, such as a round face, bilateral ptosis, large, protruding ears, short stature, and panhypopituitarism with abnormal pituitary findings on MRI. CMA confirmed a 14.9-Mb deletion at 18p11.32p11.21 (chr18: 136,227–15,099,116)×1. The 18p deletion syndrome was first described in 1963 by de Grouchy et al. [8] In addition to the typical clinical features, isolated hormone deficiencies and hypopituitarism have been described as features of 18p deletion syndrome [7,9,10]. According to the most recent review of 18p deletion syndrome [7], approximately 23% of patients had isolated GH deficiency, while 13% had hypopituitarism. However, to date, only one case of panhypopituitarism associated with 18p deletion syndrome has been reported [9]. Therefore, it is noteworthy that our patient is the second case of panhypopituitarism with pituitary hypoplasia related to 18p deletion syndrome. Pituitary abnormalities on MRI, as in our patient, are highly associated with loci of deletion in 18p11.32–18p11.22 (genomic size 9,849,183 bp) according to the 18p deletion phenotype map (https://genome.ucsc.edu). Moreover, clinical features of our patient such as round face, short stature, and ptosis are correlated to the phenotypic map proposed by Brenk et al. [3] These findings support some degree of genetic homogeneity in 18p deletion syndrome.
More than half of cases with 18p deletion syndrome have breakpoints in the centromeric region, while the remainder of the breakpoints are scattered along the entirety of the short arm [11]. Because some of these patients have a unique deletion, a high-resolution diagnosis using CMA is critical. By identifying genes involved in the deletion by CMA, it is possible to provide further information to family members about the expected manifestations and prognosis of their affected child. To date, few known functional genes on 18p have been identified. TGIF1 (3,451,591–3,458,406) related to holoprosencephaly is located in chromosome band 18p11.31. GNAL (11,689,014–11,885,683) related to dystonia is found in chromosome band 18p11.21. Our patient had breakpoints distal to 18p11.1 (18p11.32–p11.21), which is associated with mild phenotypic abnormalities and normal intelligence, while deletions in the centromeric region are known for more distinct phenotypic abnormalities such as moderate to severe intellectual disability and typical facial dysmorphism, which suggests that a critical region for intellectual disability may exist between 18p11.21 and 18p11.1 [4].
The deleted region in our pat ient ( arr 18p11.32p11.21(136,227–15,099,116)×1) encompasses 26 OMIM genes. Of these, four genes, GNAL, TUBB6, AFG3L2, and PTPN2, are dosage sensitive. Our patient did not show any dystonic features, and dystonia rarely occurs among 18p deletion patients who are heterozygous for GNAL. However, the patient may develop some additional phenotypic features of 18p deletion with age. Follow-up examinations will be necessary to see whether the patient with GNAL, AFG3L2, PTPN2, and TUBB6 deletions could eventually develop dystonia, spinocerebellar ataxia, autoimmune disease, and neurodevelopmental disorder [12-14].
The positive impact of GH therapy on linear growth in our patient is consistent with previous reports of 18p deletion syndrome [15,16]. Early diagnosis and hormone replacement therapy with levothyroxine, GH, and hydrocortisone for multiple hormone deficiencies may enable favorable growth and neurodevelopmental outcome.
In conclusion, this study identified a patient with craniofacial anomalies, short stature, pituitary hypoplasia, and ptosis as having 18p deletion syndrome using karyotyping and CMA. GH therapy was beneficial in this patient with 18p deletion syndrome. A combined pituitary hormone stimulation test was performed at the early stage of diagnosis, and deficient hormone replacement therapy was essential for improved growth rate and favorable neurodevelopmental outcome. Further studies in genotype-phenotype correlation are needed for individualized treatment strategies and prognosis prediction.
Notes
Ethical statement: This study was approved by the Institutional Review Board (IRB: 2012-05-080-008) of Samsung Medical Center and conducted according to the Declaration of Helsinki ethical principles. Parental informed consent was obtained in accordance with IRB standards.
Conflict of interest: No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was supported by a grant from Samsung Medical Center (#GFO2160061). We sincerely appreciate our patient and her family for their participation in this study.
Supplementary material
Supplementary Fig. 1 can be found via https://doi.org/10.6065/apem.2019.24.1.60.