Introduction
Adrenal hypoplasia congenita (AHC) is a rare, potentially life-threatening inherited disorder that is characterized by impaired development of the adrenal cortex. It is inherited in either an X-linked or an autosomal recessive manner. The X-linked form is caused by more than 100 different mutations or complete deletion of the NR0B1 gene (previously known as DAX1 gene) which is located on Xp21.2. This gene is expressed in tissues directly involved in steroid hormone production and reproductive function i.e., adrenals, gonads, hypothalamus and pituitary gonadotrope cells and codes for DAX-1 protein which belongs to the orphan nuclear receptor superfamily and plays a key role in gonadal and adrenal development [1].
Clinically, AHC manifests in the majority of cases as a syndrome of adrenal insufficiency in infancy or early childhood. However, a large clinical spectrum exists with some cases being diagnosed later in childhood or adolescence. Other disorders of male genitalia such as cryptorchidism or microphallia at birth have been reported [2].
Testicular microlithiasis (TM) is characterized by calcium deposits within the seminiferous tubules presenting as multiple, uniform echogenic foci scattered throughout the testicular parenchyma. It has been found to be associated with several benign conditions such as cryptorchidism and congenital adrenal hyperplasia (CAH) as well as with testicular malignancy [3].
In this study, we report a boy who was diagnosed with AHC at the age of 8 months due to failure to thrive and repeated episodes of vomiting, dehydration, hyperkalemia and hyponatremia. During his last follow-up appointment at the age of 11 years, he was diagnosed with TM. To the best of our knowledge, this is the first case of an AHC patient diagnosed with TM in the follow-up.
Case report
We retrospectively review a case of X-linked AHC. Ethical review board approval (Aristotle University of Thessaloniki Ethical Committe, A13729) and informed consent from both parents of the proband participating in this paper was obtained in accordance with the national laws.
Our patient was born at term with an Apgar score of 9 at 1 minute and with the following somatometric parameters: weight: 3,120 g (25thŌĆō50th percentile), length: 50.5 cm (25thŌĆō50th percentile), head circumference: 34.5 cm (25thŌĆō50th percentile). He was the second child of phenotypically healthy nonrelated parents (his mother is of Armenian origin and his father is Greek) with no family history of adrenal disease. His past medical history was significant for several episodes of diarrhea, vomiting, and dehydration since the age of 40 days that were treated as infections of the gastrointestinal tract. Characteristically, all of these episodes had some degree of hyperkalemia and hyponatremia without any hypoglycemia.
He was admitted to our clinic at the age of 8 months due to a 2-day history of vomiting, diarrhea and refusal to eat. On the day of admission his weight was 8,650 g (50thŌĆō75th percentile), his length was 71 cm (75th percentile) and he had low sodium (127 mmol/L), high potassium (5.96 mmol/L) and mild metabolic acidosis (pH, 7.31). Due to his clinical presentation and his past medical history, an endocrine evaluation was performed that confirmed adrenal insufficiency: cortisol levels were very low (3 ╬╝g/dL) with an elevated serum adrenocorticotropin (ACTH) level (236.0 pg/mL) and a normal 17-hydroxy-progesterone level (37 ng/dL; reference, 20ŌĆō40 ng/dL). His aldosterone (<7.0 ng/dL; reference, 7ŌĆō90 ng/dL) and renin levels (0.345 ng/mL/hr; reference, 0.5ŌĆō1.9 ng/mL/hr) were also low. A standard-dose short ACTH stimulation test was performed which confirmed the impaired cortisol response to ACTH (cortisol levels at 30 minutes, 3.07 ╬╝g/dL and at 60 minutes, 2.44 ╬╝g/dL). The patient was commenced on hydrocortisone and fludrocortisone substitution with prompt improvement in his electrolytes and his general condition.
Genomic DNA was extracted from peripheral blood lymphocytes of the patient and the molecular examination revealed a complete deletion of his NR0B1 gene, namely a deficit of 5 Mbp in Xp21.2 position. Normal levels of creatine kinase and triglycerides in our patient excluded the possibility of contiguous deletion syndrome i.e., combination of X-linked AHC with glycerol kinase deficiency and Duchenne muscular dystrophy.
The patient had normal psychomotor and language development. More specifically, he first sat at the age of 8 months, he walked alone at the age of 13 months, and he combined words at the age of 22 months. His school achievement is average without any special educational needs or learning disabilities diagnosed. His skeletal maturation rate was also average: his first deciduous tooth appeared at the age of 6 months and his first permanent tooth at the age of 5 years and 9 months. His bone age was slightly delayed during childhood: 4.5 years at chronological age of 5 years and 8 months and 6 years at chronological age of 7 years and 6 months.
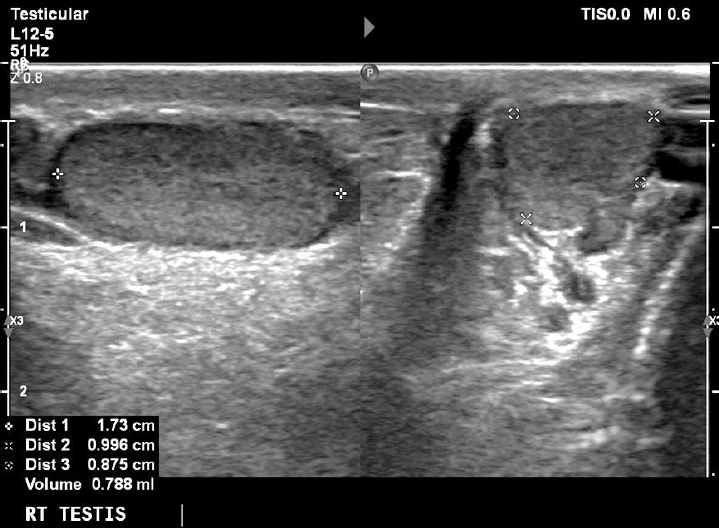
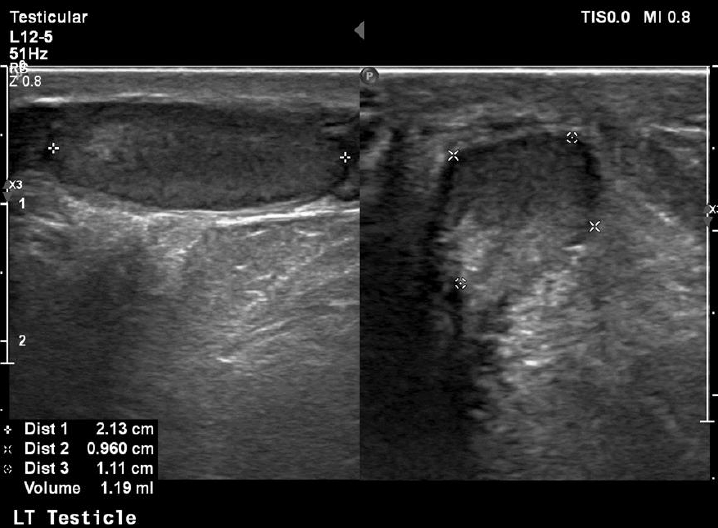
At his last follow-up visit at the age of 11 years, his bone age was 10 years and his genitals were Tanner I with testes volume <3 mL and penis length 7.5 cm (reference, 6.2┬▒1.3 cm). In order to assess his sexual maturation, a gonadotropin-releasing hormone (GnRH) stimulation test was performed which confirmed the lack of puberty onset (luteinizing hormone [LH] at 0 minute, 0.32 mIU/mL; peak LH, 2.91 mIU/mL; and peak LH<peak FSH), but of course at this early age no diagnosis of hypogonadotropic hypogonadism (HH) can be made. His inhibin B levels were 95 pg/mL (reference, 41ŌĆō328 pg/mL). An ultrasound (US) exam of his testes revealed their small size (1 and 1.2 mL, respectively) as well as classic grade 1 (5 microliths per US field) bilateral segmental TM [4]. A US exam of his kidneys revealed no calcium deposits in kidney parenchyma.
His height was 141.6 cm (25thŌĆō50th percentile) and his weight was 57 kg (>95th percentile). Despite his normal height for age and sex, due to his low height velocity (3.2 cm during last year) a glucagon provocation test was performed which excluded GH deficiency (peak GH, 12.7 ng/dL).
The patient is still on substitution therapy with hydrocortisone 7.5 mgŌĆō2.5 mgŌĆō2.5 mg orally per day (8.35 mg/m2) and fludrocortisone 0.1 mg orally once a day with good control. A follow-up visit is scheduled at 6 months.
Discussion
In this case report we describe a teenage boy with X-linked AHC who was diagnosed with TM during his regular follow-up. X-linked AHC is caused by mutations in NR0B1 gene and is predominantly a disease of early infancy with the majority of cases presenting in the first days or weeks of life with the typical clinical and biochemical findings of adrenal failure. However, the disease can be diagnosed later in childhood due to variable glucocorticoid and mineralocorticoid deficiency, in adolescence due to HH, or even in adulthood due to infertility [2]. Its estimated incidence ranges from 1:12,500 live births [5] to less than 1:70,000 males [6]. No specific populations are known to be at greater risk for the disease.
Since NR0B1 gene and its protein DAX-1 play a key role in gonadal development, it is no wonder that patients with X-linked AHC have been reported to present not only dysfunction of the hypothalamic-pituitary-gonadal axis but also disorders of gonads per se. More specifically, HH in adolescence is usually caused by a combined hypothalamic failure in GnRH release and pituitary defect in LH/FSH production [7]. Further, GnRH or gonadotropin treatments are usually inefficient to improve spermatogenesis in patients carrying NR0B1 mutations, suggesting that a primary testicular defect is also present [8]. In addition, cryptorchidism and microphallia at birth have been reported in several cases of AHC patients suggesting that NR0B1 gene might play a more general role in testes formation and function [2]. Thus far, our patient has not shown any signs of puberty but at the age of 11 years, it is too early to diagnose HH. The volume of both his testes is rather small for his age but he never had cryptorchidism and the length of his penis is within normal limits.
On the other hand, TM was first described as a US finding 30 years ago [9] while the first report of a pediatric case (as an incidental finding on X-ray) was published much earlier, in 1970 [10]. The term refers to multiple, uniformly echogenic foci less than 3 mm in diameter that can be found either diffusely or segmentally located throughout the testicular parenchyma. The prevalence of TM in boys varies between 1 and 4% [11,12]. TM is classified with respect to the number of microliths per US field as limited or classic grade 1 to 3 [4]. Our patient was diagnosed with classic grade 1 (5 microliths per US field) bilateral segmental TM (Figs. 1, 2).
The etiology of TM is not known. It has been found in children to be associated with several benign conditions such as varicoceles, cryptorchidism and orchidopexy, scrotal trauma, testicular torsion, CAH as well as Down and Klinefelter syndromes [11,13]. Further, in CAH patients it has been found to coexist with testicular adrenal rest tumors which, if left untreated, may lead to testicular structural damage and infertility [3,14]. Although the above mentioned associations have not been proven to be causal, they may indicate that TM is a predictor of future testicular disorders. Our patient did not have any of the testicular pathologies already described to coexist with TM.
In addition, TM association with infertility has been reported in some studies. Low tubular diameter, tubular degeneration and obstructing intratubular debris in seminiferous tubules could explain the possible association [15]. Since X-linked AHC has been shown to be frequently associated with infertility in adult life, our patient may have another factor, namely TM, predisposing him to future infertility. For the time being, no final conclusion regarding his fertility can be drawn since no semen analysis can be performed and his inhibin B levels are somewhat low but within normal limits [16].
Further to its association with benign conditions, TM in adults has been linked to testicular malignancy [17,18]. In children, only 5 cases of testicular malignancies have been described in patients known to have TM. Cases of testicular tumor development in boys with TM prospectively followed-up are even rarer [12]. X-linked AHC has not been linked with increased risk of malignancy. It thus seems improbable for our patient to be at increased risk of developing malignant testicular tumor in the future either because of his primary disease or because of the presence of TM. Nevertheless, a close follow-up is warranted.
As is the case with many diseases that can be diagnosed together with TM, we cannot be certain if the coexistence of X-linked AHC and TM in our patient is coincidental or a cause-and-effect phenomenon. Furthermore, since AHC is a very rare disease it is quite difficult to identify the true prevalence of TM in AHC patients. Nevertheless, since TM has been associated with several benign and possibly some malignant conditions in children, a careful follow-up of our patient both for his primary disease as well as for the evolution of TM is required. Even if there is no consensus regarding the best follow-up of such patients, a prudent approach would include frequent self-palpation of the testes by the patient, clinical and laboratory examination every 6 months (both for dose adjustment, for possible puberty induction and for TM), as well as a US examination of the testes at least once a year. No testicular biopsy or tumor markers are required diagnostically at the time being [12,19].
In conclusion, this is the first case ever reported of an X-linked AHC patient diagnosed with TM in the follow-up. Since X-linked AHC is a rather rare disease, it is difficult to confirm this finding in large patient populations. Nevertheless, clinicians caring for such patients should bare this possibility in mind and should perform a US of the testes both to assess their volume progression and to timely identify any associated conditions such as TM.