Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by the deficiency of enzymes required for the conversion of cholesterol to glucocorticoids, mineralocorticoids, and adrenal androgens [1]. 21-hydroxylase deficiency (21-OHD) CAH (OMIM #201910) accounts for >90% of all cases, with an incidence rate of approximately 1 in every 10,000 to 20,000 live births, for all forms of CAH [2].
It is caused by homozygous, or compound heterozygous, mutations in CYP21A2, on chromosome 6p21.3 [3]. CAH can be broadly dived into early onset classic CAH and late onset nonclassical CAH (NC CAH).
The late-onset phenotype (NC CAH) is characterized by normal serum cortisol and aldosterone levels and absence of early childhood virilization. Enzyme activity in this form ranges from 10% to 40%. Biochemically, variable degrees of hyperandrogenemia are present; however they most often do not cause symptoms until late childhood or adolescence, with many patients, particularly males, remaining asymptomatic throughout their lives [1].
Biochemical testing is usually the initial diagnostic step in the workup of individuals suspected of having CAH, with 17 hydroxyprogesterone levels being elevated in all forms of 21-OHD. Although the different forms of 21-OHD are distinguished on clinical grounds, molecular testing is being used more often, especially in cases picked up via newborn screening before the clinical phenotype is evident, or where biochemical testing is ambiguous [4]. Here we present 2 individuals in a family who underwent long-term treatment for classic CAH, before revaluation with molecular testing revealed NC CAH not requiring treatment.
Case report
This research related to human use has been complied with all the relevant national regulations, institutional policies and in accordance with the tenants of the Helsinki declaration, and has been approved by the author's or equivalent committee. Informed consent was obtained from the parents of the patients.
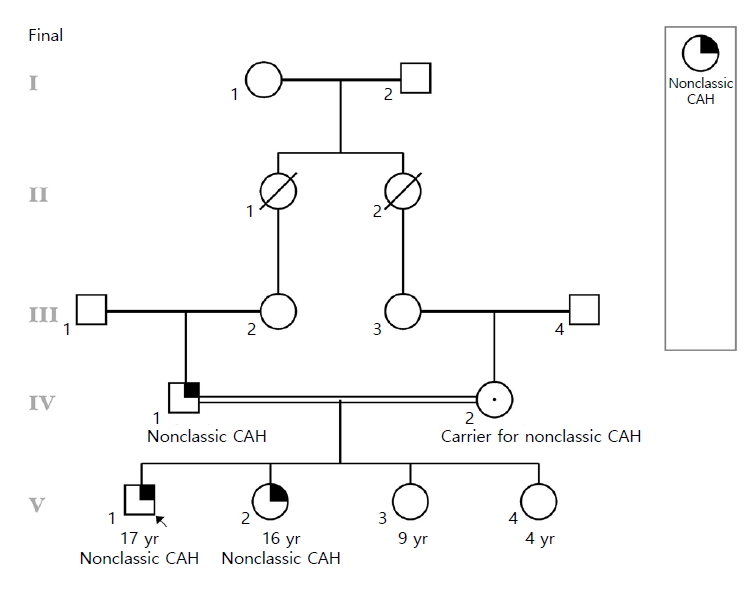
The proband was a male born to consanguineous parents (Fig. 1). He had an acute viral fever at 4 years of age, and was found to have mild hyponatremia and hyperkalemia (sodium 131 mEq/L and potassium 6.1 mEq/L, respectively). Further investigation revealed increased 17-hydroxyprogesterone (17-OHP) and adrenocorticotropic hormone (ACTH) levels (Table 1), and a suboptimal rise in cortisol levels following ACTH1-24 stimulation (13.4 ö¥g/dL after 60 minutes). At the time of diagnosis proband was of normal growth and stature (height=107 cm, z-score=-0.4, 32nd percentile; weight=20.8 kg, z-score=+0.8, 80th percentile; body mass index [BMI]=18.2 kg/m2). Treatment was thus begun for presumed salt wasting (SW) CAH using replacement doses of hydrocortisone and fludrocortisone. He had normal male genitalia, descended testes, and no evidence of precocious adrenarche or puberty. He was seen by various providers sporadically over the years, and finally was reviewed in our pediatric endocrinology clinic at 17 years of age. He was on subphysiologic hydrocortisone replacement (7.7 mg/m2/day) as well as fludrocortisone (0.2 mg per day), without any symptoms, no history of prostration with illness, and normally-timed pubertal changes. He was obese with normal stature (height=170 cm, z-score=-0.8, 21st percentile; weight=110 kg, z-score=+2.4, 99th percentile; BMI=38.1 kg/m2), and had normal testes with Tanner stage V pubic hair.
After his diagnosis, the 3-year-old sister was screened for increased 17-OHP and ACTH levels, which were found to be mildly elevated (Table 2). At the time of her diagnosis she was also of normal growth and stature (height=94 cm, z-score=-0.05, 48th percentile; weight=14 kg, z-score=0.03, 51st percentile; BMI=15.8 kg/m2). She was also started on physiologic doses of hydrocortisone after ACTH1-24 -stimulation showed suboptimal rise in cortisol levels (13.12 ö¥g/dL after 60 minutes). She also presented to us in the Pediatric Endocrinology clinic at the age of 15 and 11 months, on subphysiologic hydrocortisone replacement (8.33 g/m2/day), with no history of virilization, precocious puberty or prostration with illness. She was obese (height=158 m, z-score=-0.7, 23rd percentile; weight=73.6 kg, z-score=1.4, 92nd percentile; BMI=29.5 kg/m2), with no hirsutism and normal female genitalia (Tanner V breasts and pubic hair).
We questioned the diagnosis of SW CAH, since they were both asymptomatic on subphysiologic hydrocortisone replacement. Work up revealed normal serum electrolytes, mildly increased ACTH and stimulated 17-OHP levels, which when combined with the lack of any symptoms, led us to suspect NC CAH (Tables 1, 2).Thus samples were taken from the family and sent for molecular testing.
Participants were tested for the presence of mutation(s) within the CYP21A2 gene and large rearrangements between CYP21A2 and the CYP21A1P pseudogene. The method used was sequencing and multiplex ligation-dependent probe amplification.
Molecular testing revealed that the proband had one deleted copy of CYP21A2, whereas the other copy harbored a likely pathogenic sequence variant (c.748C>T, p.R124C). The p.R124C variant has been reported previously in an individual with a diagnosis of CAH [5]. Previous functional studies indicated that this variant has approximately 16% residual activity [5]. The arginine (R) at this position is highly conserved across species and there is a large physicochemical difference between arginine (R) and cysteine (C) [6]. In silico analysis, based on SIFT, PolyPhen-2, MutationTaster, and Align GVGD suggests that this variant might affect protein function based on the amino acid change. SIFT categorized these changes as damaging. PolyPhen-2 scored it as probably damaging with HumDiv and HumVar scores of 1.000 and 0.997, respectively (scale: 0.000 ã not damaging ã to 1.000 ã damaging). MutationTaster classified it as "disease causing" with a score of 180 (scale: 0.0 ã not disease causing ã to 215 ã disease causing). Finally, AlignGVGD gave it a GD score of 70.97, which puts it into the most "likely damaging" prediction Class C65 (scale: Class C0 ã not damaging ã to C65 ã most likely to be damaging) [6,7]. Taken together, these findings suggested that p.R124C is likely pathogenic. If p.R124C is pathogenic, the most likely phenotype due to compound heterozygosity for p.R124C and a large deletion would be NC CAH.
The probandãs sister had the same genotype. To our surprise, family studies indicated that the father had the same genotype as the 2 children, with one copy deleted and the other harboring the p.R124C mutation. The mother was heterozygous for the large deletion. Therefore, a diagnosis of NC CAH was confirmed, and consequently, we discontinued the current treatment regimen, with regular follow-up visits. Both siblings remain asymptomatic without medication, with mild, expected laboratory derangements, probandãs sister had a 17-OHP levels (9,072 ng/dL) 1 year after the cessation of medication. Follow-up testing could not be performed on the proband.
Discussion
Classic CAH and NC CAH differ widely in their phenotypes, and thus it is important for clinicians to be able to differentiate between the two.
The diagnosis of CAH is primarily based upon the history, physical examination and laboratory values of 17-OHP. The Endocrine Society has outlined the diagnostic criteria in its consensus statement to aid physicians in the process when evaluating patients for 21-OH CAH [2]. A useful diagnostic cutoff for classic CAH is a stimulated 17-OHP level >10,000 ng/dL, 1,000ã10,000 ng/dL for NC CAH and below 1,666 ng/dL for those not affected; this last category will include heterozygotes as well as some normal individuals (particularly those who are physically stressed, e.g., preterm babies). In addition, an ACTH-stimulation test and corresponding 17-OHP values are used to rule in- or rule out disease [2]. However, where the above testing inconclusive, such as in this case where the 17-OHP was in the range of classic CAH in the brother despite a lack of symptoms, molecular testing can be helpful in revealing the underlying defect [3].
As mentioned earlier, NC CAH is due to a mutation and/or deletion of the CYPA21A2 gene that leaves the affected individual with a total enzyme activity that is less than 50% of a normal person, but larger than about 10%. More severe functional impairment is associated with classic CAH.
CYP21A2 and its associated pseudogene(s) (CYP21A1P) are located on chromosome 6 within the HLA locus, the most highly recombinant portion of the human genome. Not surprisingly, this leads to frequent genomic rearrangements between CYP21A2 and its highly homologous pseudogene, CYP21A1P. These events might create large deletions (involving parts or all of CYP21A2 and CYP21A1P, or only CYP21A2), recombinant hybrids of gene and pseudogene, or microgene conversions of CYP21A2, where small portions of CYP21A1P get transferred into CYP21A2 [4].
Guidelines set out by the Endocrine Society dictate that classic CAH be a treated with mineralocorticoids and sodium chloride supplements in infancy and Hydrocortisone should be given as maintenance therapy. While, symptomatic patients with NC CAH should receive the same treatment as their CAH counterparts, asymptomatic patients should not receive any treatment. Unwarranted diagnosis and treatment not only burdens the healthcare infrastructure, but is bothersome for the patients even though they are physically healthy.
To conclude, asymptomatic patients with NC CAH do not require aggressive treatment and repetitive diagnostic testing is unhelpful [8]. Genetic evaluation is important as it identifies the specific defect and is helpful when the diagnosis is uncertain. In addition, it is also vital to screen the family with genetic testing as it will assist in genetic counseling for future offspring and allow identification of asymptomatic carriers.