Introduction
Gonadal differentiations arise from the sex chromosome determined at the beginning of gestation [1]. The existence of the Y chromosome plays a key role in the development of fetal gonads into the status of male sex determination, and is essential for spermatogenesis. In addition, the sex-determining region Y (SRY) is a well-known gene as one of the testis-determining factors located in the distal proximal region of the short arm of the Y chromosome [2]. Structural aberrations of the Y chromosome are reported in approximately less than 1% of newborn male babies. They appear at a breakage point and include various types of abnormalities, such as deletion, duplication, translocation, ring chromosomes, and isodicentricchromosomes [3]. The karyotype of the 46,X,+mar is one example of males having testicles without the normal structural Y chromosome in their non-mosaic karyotypes, which is rarely reported (Table 1) [2,4,5].
A marker chromosome (mar) is a structurally abnormal chromosome that has no part that can be identified via conventional cytogenetic karyotype analysis [6]. The incidence of marker chromosomes in the perinatal period varies from 4%–15% [6] and 7.9% in the referred population [7]. The importance of a marker chromosome depends on what genetic information and materials are preserved within it. It is critical to clarify the characteristics and origin of the marker chromosome because they are related to the phenotype and pathologic phenomenon.
When a sex chromosome is not described in a conventional karyotype-carrying marker chromosome, it is generally assumed to be sex chromosome component within the marker chromosome. In this case, the fluorescence in situ hybridization (FISH) technique is conducted on a preferential basis to verify the sex chromosome.
Twenty-seven genes have been confirmed in the Y chromosome. Nine genes are located in the short arm, and 18 genes are in the long arm of the Y chromosome [8]. Deletions on Yq are related to growth alterations, skeletal development, spermatogenesis, and fertility. Therefore, cytogenetic molecular techniques are required to clarify the genetic and clinical characteristics of the marker chromosome.
Herein, we report a boy presenting gynecomastia and short stature carrying 46,X,+mar, which originated from the Y chromosome, and microdeletions were identified on Yq using FISH, polymerase chain reaction, and direct sequencing with a literature review.
Case report
A 15-year-old boy was referred to the pediatric endocrinology service due to his persistent gynecomastia over 2 years and short stature. He was the first boy of two brothers from nonconsanguineous Korean parents. His birth weight was 2,800 g after a full-term delivery. He has been followed up in pediatric psychology because of mild intellectual disability (intelligence quotient, 71). His height, weight, and body mass index were 157.4 cm (5th–10th percentile), 62.1 kg (50th–75th percentile), and 25.06 kg/m2 (85th–90th percentile), respectively. His father's height was 174 cm, his mother's height was 159 cm, and the midparental height was 173 cm. His growth velocity was 4 cm/yr, and his parents reported an indistinct pubertal spurt. He manifested nontender bilateral breast enlargement. A small phallus was noticed, and the bilateral testes were palpated. The volume of each was measured as 6 mL. He was at the pubertal stages of developing the breasts of Tanner III and the pubic hair of Tanner III. A knuckle-dimple sign was shown on his left hand, and the fourth toe of his left foot was remarkably short. A basal hormonal study showed elevated estradiol and follicle-stimulating hormone (FSH) levels, and his testosterone (T) concentration was within the normal range (Table 2). The normal pubertal status of luteinizing hormone (LH) responses was recognized, however, an increased basal FSH level was revealed during a provocation test (Table 3).
Radiographs showed short fourth and fifth metacarpal and fourth metatarsal bones. His bone age was 15 years old by the Greulich and Pyle method at a chronological age of 15 years (Fig. 1). An abdominopelvic ultrasound revealed no ovarylike structure or uterus and normal kidneys. Normal testes and a varicocele in the left testis were demonstrated; however, the 4-year-followed testicular volume decreased to 3.8 mL and 3.9 mL in each testis according to testicular ultrasound. The sella magnetic resonance imaging results was normal.
1. Cytogenetic and FISH analysis
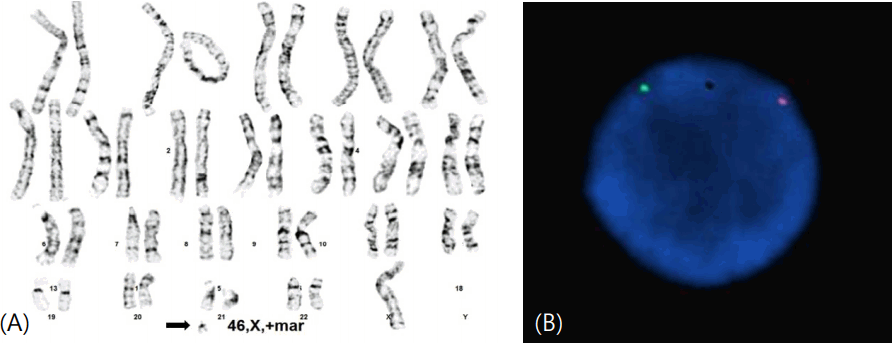
Phytohemagglutinin-treated peripheral blood lymphocytes were prepared and cells were cultured, and then harvested after 72 hours following standard protocol. Giemsa dye mixture was used for conventional G-bands by trypsin using Giemsa (GTG) technique. Then metaphase cells were analyzed to reported as 46,X,+mar by the International System for Human Cytogenetic Nomenclature (ISCN 2009) (Fig. 2A). The karyotypes of family members were normal.
FISH analysis was conducted to identify the existence of Y chromosome on marker chromosome. AneuVysion Test Kit (Abbott Laboratories, Abbott Park, IL, USA) was utilized according to manufacturer's instruction. Vysis CEP Y, orangecolored probes for DYZ3 corresponding centromere of the Y chromosome (Yp11.1–q11.1) and Vysis CEP X, greencolored probes for DXZ1 corresponding centromere of the X chromosome (Xp11.1–q11.1) were hybridized to metaphase cells. Figures of images were obtained with a CytoVision platform (Leica Biosystems, Richmond, IL, USA). FISH study revealed that the marker chromosome in this case includes the centromere of the Y chromosome of all 150 cells in metaphase (Fig. 2B).
2. Polymerase chain reaction and Y-specific STS
Genomic DNA of patient was extracted from peripheral blood samples. QIAamp DNA Blood Midi Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Extracted DNA were stored at -70℃ until experiment. Twenty-five Y chromosome specific sequences located in both arms of the Y chromosome were used to detect cryptic Y chromosome material by polymerase chain reaction (PCR) analysis. All primers were designed by Primer3 program (http://bioinfo.ut.ee/primer3/). The Taq polymerase reagents were purchased from Takara (Takara Bio, Otsu, Japan). PCR was performed with 20 ml of PCR amplification reaction mixture containing 10X Ex Taq DNA polymerase buffer with 2.5 mM of each dNTP, 0.5 mM of each primer, 500 ng of DNA, and 2.5 units of Taq DNA polymerase.
Amplifications were conducted in duplicate with the PTC-200 thermocycler (MJ Research) with the following profile: 94℃ for 5 minutes, 35 cycles of 95℃ for 30 seconds, 50℃ for 30 seconds, 72℃ for 30 seconds and final extension at 72℃ for 10 minutes. One negative and positive sample was loaded to compare with the expression patterns of the patient. One percent (1%) agarose gels were used to separate PCR products by electrophoresis.
Seven of Y-specific sequence-tagged site (STS): SRY, proximal border of pseudoautosomal region of Y (PABY), DYS14, amelogenin sequence of Y (AMGY), sY57, DYS265 were confirmed on Yp and centromere (DYZ3) was verified using PCR and direct sequencing (not shown). Azoospermia factor (AZF) b region was partially deleted, and AZFa and AZFc were completely deleted. Two STS probes of sY143 and Y chromosome RNA recognition motif (YRRM) in AZFb showed positive signals corresponding to Yq11.223 (Fig. 3).
Therefore, the karyotype of this case was interpreted as 46,X,der(Y)del(Y)(q11.21q11.222) del(Y)(q11.23qter).
Discussion
Physiologic pubertal gynecomastia is common in adolescents at 13 or 14 years of age and generally manifests a unilateral or bilateral, transiently tender, rubbery or firm mass in a concentric location. Most physiologic pubertal gynecomastia regresses within 1–2 years [9]. A cohort study reported that if it is persistent beyond 2 years and prominent over B3, endocrine evaluation should be considered [10]. The persistent, prominent gynecomastia group has underlying causes, including increased aromatase activity, adrenal or testicular neoplasm, partial androgen insensitivity syndrome, and a disorder of sex differentiation (DSD), such as Klinefelter syndrome, 46,XX, testicular (SRY+)- associated hypogonadism, or genital abnormalities [11]. When gonadotropin levels are elevated, the karyotype should be examined [9]. Therefore, the present case manifested clinical features suggesting DSD, such as a small phallus and small testicles, so karyotyping was performed. Interestingly, his karyotype of 46,X,+mar was identified as 46,X,der(Y).ish der(Y) (DAZ3+). In addition, deletions of Yq and preserved SRY were verified by PCR.
This present case carrying 46,X,+mar showed obesity, short stature, small testes, short fourth, and fifth metacarpals and metatarsal bones. These are similar clinical manifestations to those in previous reports (Table 1). 46,X,del(Yq) is associated with tubular hyalinization, spermatogenic maturation arrest, Sertoli cell-only syndrome (SCOS), and small testes with testicular lesion, and related comorbidities are short stature, intellectual disability, and gynecomastia [12].
The short arm of the Y chromosome is subdivided into intervals 1 to 4A [13], and SRY is a key factor for sex determination located on distal PABY of Yp. Testis transcript Y1, testis transcript Y2, and AMGY are located on interval 3 of Yp [11]. In this case, the existence of SRY was confirmed by PCR, which explains his male phenotype.
The long arm of the Y chromosome was subdivided into intervals 4B to 7. Polymorphism of the heterochromatin located in Yq12 at interval 7 is frequently observed; however, its function is inert. In contrast, the Yq11 region is known to influence growth control, teeth development, the anti-Turner effect, and fertility [13,14]. Y-specific growth control regions (GCY) are related to short stature [15] and located in interval 5 within a narrow range immediately below the centromere. GCY is speculated to control growth hormone and the insulin-like growth factor-I receptor or postreceptor pathway [15].
Various combinations of manifestations, such as short stature, a webbed neck, cubitus valgus, short fourth and fifth metacarpals, a low posterior hairline, pigmented nevi, and gynecomastia, are related to microdeletions of the Yq chromosome in some cases [4,5,14]. In the present case, the male patient showed skeletal deformities of the hand and foot, short stature, and gynecomastia with no DNA loci among sY82, sY83, sY84, DYS7, and DYS280 in interval 5 on proximal Yq. These findings are consistent with previous reports regarding the correlation between proximal Yq deletion and skeletal deformities [16]. Many reports of Yq microdeletions have been performed in adult age during the evaluation of fertility; however, pubertal gynecomastia is first described in this case.
Structural microdeletions of the Y chromosome are found in 7.7% of infertile Korean adults and vary between 5.7%–21% worldwide [3]. In addition, Yq microdeletion can be attributable to varicoceles, cryptorchidism, and infertility as well as intellectual disability [12]. In this case, the patient had a varicocele in 1 testis and was suffering from mild intellectual disability.
The AZF within the Yq region is related to infertility and subdivided into AZFa, AZFb, and AZFc. Deleted AZF is associated with oligospermia or severe azoospermia. The deletion of the AZFc region occurs most frequently, followed by AZFb, whereas the complete deletion of AZFa is rarely discovered in male infertility. In this case, YRRM and DYS231 (sY143) were detected from several loci of Yq11.223 in the AZFb region. This is consistent with partial deletion of AZFb in interval 6. However, no loci were identified in the AZFa and AZFc regions. In addition, the complete deletion of AZFa causes small testes and SCOS. SCOS is a condition of spermatogenic failure in seminiferous tubules lined with only Sertoli cells confirmed by testicular biopsy, and azoospermia should be accompanied by semen analysis. In general, SCOS is diagnosed in adults while examining sterility. Typical laboratory findings are normal T, mainly normal or elevated LH, decreased inhibin B, and increased FSH 2–3 times [3]. Inhibin B and FSH are useful predictors or markers of Sertoli cell function during a follow-up period [17]. In this case, testicular biopsy and sperm analysis were not performed due to the patient's young age. However, 4 years later, both testicular volumes were remarkably reduced from 6 mL to less than 4 mL. Hormonal concentrations of T were found within the normal range as well as 4 years later, the FSH level was markedly elevated, and complete deletion of AZFa was confirmed by PCR. These findings suggest the possibility of SCOS. Even though men are presumed with SCOS, sperm retrieval could be possible via testicular sperm extraction (TESE) or micro-TESE, which appear to be recommendable in such cases [18]. Moreover, several studies have been reported that it is possible to identify sperm cells in TESE in 57% of partial deletion of AZFb, and 43% of partial deletion of AZFb-c, but, in 7% of complete deletion of AZFb and 3% of complete deletion of AZFb-c [19]. Judging from the partial deletion of AZFb, although this patient received neither sperm analysis nor TESE yet, the possibility of sperm retrieval remains to be present. Therefore, it is clinically important to verify the microdeletions of Yq to determine the treatment and prognosis.
In conclusion, when there is gynecomastia accompanied by raised gonadotropin levels, karyotyping should be performed. Furthermore, when a conventional banding technique demonstrates a marker chromosome instead of a normal sex chromosome, it is necessary to conduct full endocrine evaluations that include cytogenetic and DNA molecular analyses to investigate the existence of a sex chromosome or SRY and the structural aberrations or breakpoints of the marker chromosome.