Introduction
Recently, a shift in pubertal timing has been reported worldwide [1]. Historical data from several countries shows that the average menarcheal age has decreased remarkably [2]. A recent study suggests similar decreases in average menarcheal age in Korean girls [3]. Consequently, the prevalence of precocious puberty has increased in Korea and it is becoming a social concern [4].
Various factors appear to affect the timing of pubertal development, including the environment and nutrition. Adequate nutrition is important for normal timing of pubertal development, and poor childhood nutrition delays growth and pubertal onset [5]. The increase of precocious puberty in girls has been coincided by increases in childhood obesity [6]. Insulin resistance is associated with obesity and is a key risk factor for type 2 diabetes mellitus, cardiovascular disease, and dyslipidemia in adulthood [7]. Early pubertal timing in girls may be associated with early insulin resistance and cardiometabolic diseases in adults [8].
However, the relationship between obesity for precocious puberty remains a controversial topic. Bordini et al. [9] suggested that excess weight, in the absence of excess sex steroid, may subtly suppress hypothalamic-pituitary-gonadal function in premenarcheal pubertal girls. Jasik and Lustig [10] reported that there is little support for an association between weight gain and early menarche.
Some groups have studied the relation of central precocious puberty (CPP) and obesity, particularly insulin resistance, but clinical studies are still lacking [11,12]. The purpose of this study was to evaluate the influence of obesity in girls with CPP. We analyzed differences in pubertal characteristics according to body mass index (BMI) and the correlation between insulin resistance and biochemical characteristics in girls with CPP. Furthermore, we investigated the association between pubertal characteristics including bone age (BA) advancement and insulin resistance.
Materials and methods
1. Study population and design
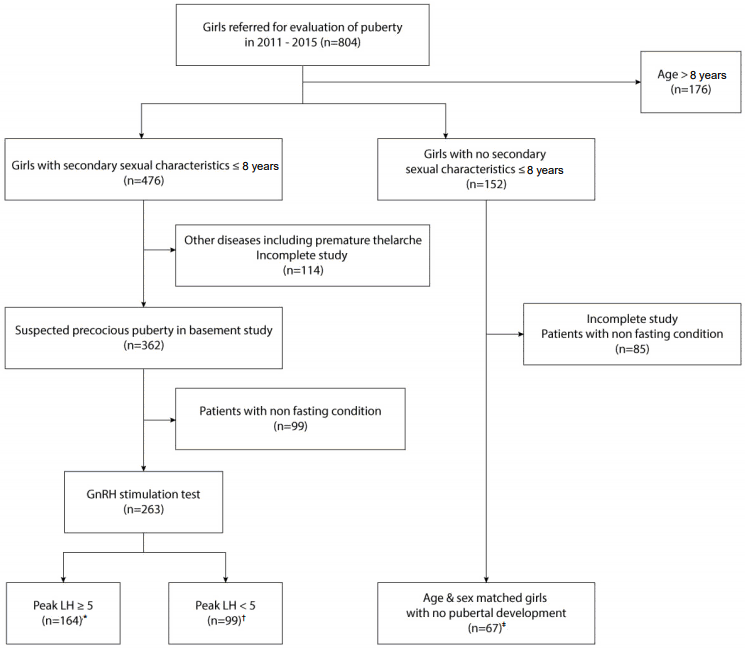
We retrospectively reviewed the medical records of 804 girls referred for evaluation of puberty from 2011 to 2015. We included girls younger than 8 years with objective breast budding above Tanner stage II [13]. We reviewed gonadotropin releasing hormone (GnRH) stimulation test results and defined CPP as peak luteinizing hormone (LH)≥5 IU/L and non-CPP as peak LH<5 IU/L. We excluded patients with tests collected under nonfasting condition, incomplete sex hormone studies, and history of any other systemic diseases. Therefore, 164 girls were included in the CPP group, and 99 girls were included in the non-CPP group. The control group comprised age- and sexmatched healthy girls (n=67) who did not have objective breast budding, that is, Tanner stage I. The study design is shown in Fig. 1. This study was approved by the Institutional Review Board of Yonsei University Severance Hospital (approval number: 3-2015-0336). Written informed consent was obtained from all patients.
2. Clinical and laboratory data
BA was evaluated by a single pediatric radiologist measured by 6 months scale with Greulich and Pyle method. BMI was calculated as weight divided by height squared. The data for height standard deviation scores (SDS), weight SDS, and BMI SDS were calculated using z-scores from the 2005 Korean National Growth Chart [14].
The hypothalamic-pituitary-gonadal axis was determined using GnRH stimulation test results. Basal serum samples were obtained before GnRH injection (100-μg Relefact; Sanofi-Aventis, Paris, France). Serum LH and follicle-stimulating hormone levels were measured using a chemiluminescence immunoassay with an analytical sensitivity of 0.2 IU/L (Beckman Coulter, Brea, CA, USA). Estradiol (E2) levels were determined using an electrochemiluminescence immunoassay with an analytical sensitivity of 5 pg/mL (Roche, Basel, Switzerland). Sex hormone binding globulin (SHBG) was determined using an electrochemiluminescence immunoassay (Roche, Basel, Switzerland). Free estradiol index (FEI) was calculated by dividing the serum E2 level (pg/mL) by the serum SHBG level (nmol/L) and multiplying by 100. The molar ratio of E2/SHBG was calculated to convert E2 to picomolar, the picogram per milliliter value is multiplied by 3.67. A peak LH of at least 5 IU/L was used as the cutoff criteria for pubertal response for the GnRH stimulation test. Insulin-like growth factor-1 (IGF-1) was measured using a solid-phase, enzyme-labeled, 2-site chemiluminescent immunometric assay (Siemens, Munich, Germany). We reviewed medical records to obtain total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, triglyceride, fasting serum glucose, alanine aminotransferase, and insulin levels. These tests were performed in a fasting state over 8 hours. Insulin resistance was assessed from fasting plasma glucose (G0, in mg/dL) and fasting plasma insulin (I0, mU/L) levels using the homeostasis model assessment-insulin resistance (HOMA-IR) and quantitative insulin sensitivity check index (QUICKI) models. HOMA-IR was calculated as I0 × G0/405 [15], and QUICKI was calculated as 1/(logG0 + logI0) [16].
3. Statistical analysis
We analyzed data with SAS ver. 9.4 (SAS Institute Inc., Cary, NC, USA). We used independent-samples T-test and analysis of variance (ANOVA) to compare the clinical and biochemical characteristics between the CPP, the non-CPP, and the control groups. We used ANOVA to compare the characteristics in the groups categorized according to BMI scores. We used a trend test to determine the significance of the increase or decrease in the variables as the increasing BMI. Pearson correlation analysis was used to define the correlation between each characteristic and insulin resistance. Multiple logistic regression analysis was used to determine the correlation between insulin resistance parameters and BA advancement. We suggested 2 models adjusted for associated variables such as age and BMI. P-values less than 0.05 were considered statistically significant. Multiple comparisons were adjusted by Bonferroni correction.
Results
1. The comparison of the baseline clinical, biochemical characteristics and insulin resistance parameters of CPP, non-CPP, and control groups
We compared the baseline characteristics of the CPP, non-CPP, and control groups (Table 1). BA and baseline serum LH, E2, FEI, IGF-1, and insulin levels were higher in the CPP group than in the control group. The HOMA-IR was higher in the CPP group than in the control group (P=0.002). The QUICKI was lower in the CPP group than in the control group; therefore, this parameter indicated high insulin resistance in the CPP group (P=0.003).
2. Baseline clinical, biochemical characteristics and insulin resistance parameters according to BMI in CPP group
We categorized the girls with CPP into 3 groups according to BMI (BMI<50th percentile, 50th percentile≤BMI<85th percentile, and BMI≥85th percentile) and compared clinical and biochemical characteristics. BA, BA advancement, FEI, alanine aminotransferase, insulin, and HOMA-IR were significantly higher in girls with high BMI than in girls with low BMI. Each of the variables showed an increasing trend with higher BMI. SHBG and QUICKI were lower in girls with high BMI than in girls with low BMI. Each of the variables showed a decreasing trend with higher BMI (Table 2).
3. The correlation between clinical, biochemical characteristics and insulin resistance parameters in total girls
We analyzed all girls including CPP, non-CPP, and control groups. Pearson correlation analysis results for HOMA-IR and QUICKI for all of girls showed that HOMA-IR was positively correlated with BA advancement, BMI SDS, FEI, IGF-1, and triglycerides and was negatively correlated with SHBG. QUICKI was negatively correlated with BA advancement, BMI-SDS, baseline LH, and FEI and was positively correlated with SHBG (Table 3).
4. The change of BA advancement associated with insulin resistance
We used logistic regression analysis to determine the probability of precocious puberty according to changes in the insulin resistance parameters. We also analyzed all girls to clarify the relationship between insulin resistance and BA advancement. When HOMA-IR increased by 1, the odds for BA advancement increased 119% (P=0.033). We analyzed the odds after adjusting for chronological age (model 1) and both chronological age and BMI (model 2) to exclude the influence of them. Similar results were achieved through models 1 and 2. When QUICKI increases by 0.01, the odds for BA advancement increased 92% (P=0.030). After adjusting for age and both age and BMI, the odds were similar but not significant (Table 4).
Discussion
Trends toward early pubertal timing have coincided with increased obesity prevalence [17]. Known factors that affect timing of puberty include estrogen in food, cosmetics, and hair care products, endocrine disrupters in the environment, nutrition and obesity [18-20]. However, these factors remain controversial. For example, while there are several studies associating weight gain and obesity with earlier thelarche, there is not much support for the association between weight gain or obesity and earlier menarche [10].
In our study, we analyzed the association between characteristics of precocious puberty and insulin resistance. Baseline serum LH, E2, FEI, IGF-1, BA, insulin levels, and insulin resistance parameters were higher in the CPP group than in the control group. Although BMI SDS did not differ among the groups, insulin resistance parameters differed in the CPP and control groups. Because one of the roles of insulin is stimulating ovary and adrenal steroidogenesis, E2 and FEI levels might increase in girls with precocious puberty [21].
Another factor that modulates maturational timing is the GH/IGF-1 axis. The GH/IGF-1 axis is affected by glucose homeostasis [22]. Sørensen et al. [23] showed that IGF-1 levels were significantly positively associated with insulin levels. While low IGF-1 serum concentrations are generally related to insulin resistance [24], we found that insulin resistance and IGF-1 were significantly higher in the CPP group than in the control group. Due to limitation of the cross-sectional design of our study, a causal relationship between IGF-1 levels, insulin resistance, and pubertal timing could not be determined. However, we found a significant relationship between IGF-1 and insulin levels in girls with CPP.
In our study, girls with high BMI had increased BA, BA advancement, FEI, insulin levels, and HOMA-IR and lower SHBG and QUICKI than girls with low BMI. Because BA advancement and increased FEI are characteristic of sexual precocity, our data suggest that obesity is associated with sexual precocity. SHBG, a glycoprotein that binds sex hormones, also links obesity and precocious puberty [25]. Hyperinsulinemia from obesity-related insulin resistance can reduce hepatic SHBG production and increase sex steroid bioavailability [26]. In our study, insulin resistance was positively correlated with BA advancement and was negatively correlated with SHBG. We found that serum insulin levels were higher in girls with precocious puberty than in girls in the control group. High levels of insulin predict adverse cardiovascular risk factors and metabolic syndrome. Insulin resistance in precocious puberty also might be associated with increased cardiovascular morbidity and mortality in adulthood [8]. Obviously, it is important to prevent obesity and insulin resistance in children.
Obesity during childhood may lead to thelarche in girls [10]. It remains unclear whether early thelarche in overweight girls is related to central activation of the GnRH-gonadotropin axis [27]. Our findings show that girls with precocious puberty had high insulin resistance. Although we did not find a direct mechanism of GnRH-gonadotropin axis activation, the association between obesity and precocious puberty is important.
One study suggested that obesity can lead to precocious puberty through leptin-induced pubertal development. Leptin serves as a signal to the hypothalamus regarding energy stores in adipose tissue and may directly stimulate GnRH and gonadotropin secretion [28]. Insulin and, paradoxically, obesity increase leptin levels [29]. Obesity-related insulin resistance with compensatory hyperinsulinemia increase serum leptin levels and may also play a role in early pubertal development. Further research is needed to identify the relationship between serum leptin levels and BA advancement.
BA studies can evaluate the rate of skeletal maturation, which can help diagnose conditions that delay or accelerate physical growth and development. Advanced BA is associated with earlier puberty [30]. We predicted the probability of precocious puberty through the quantification of insulin resistance and BA advancement. When insulin resistance increases, BA advancement occurs, which may results in precocious puberty. Our findings show that the risk of BA advancement increased 119% when HOMA-IR increased by 1. Although there are multiple factors influencing precocious puberty, our results suggest that insulin resistance may be an important factor for CPP after adjusting for age and BMI.
There was a limitation in our study to classify CPP and non-CPP with peak LH. We defined CPP as peak LH≥5 IU/L and non-CPP as peak LH<5 IU/L. Accordingly, non-CPP group includes peripheral precocious puberty and slowly progressive CPP. In addition, the control group comprised girls who did not have secondary sexual character among the girls visited the hospital to check their puberty status. It is attributed to a hospital based observation study.
In conclusion, our findings suggest that insulin resistance is elevated and associated with BA advancement in girls with CPP. Unlike other studies, our retrospective study included more than 100 patients with CPP from a single country. Longitudinal prospective studies that include more precise methods of evaluating body fat mass, such as triceps skin-fold thickness or waist circumference, are needed to determine the causality between obesity and precocious puberty.