Effects of growth hormone on glucose metabolism and insulin resistance in human
Article information

Abstract
Growth hormone (GH) is important for promotion of somatic growth and the regulation of substrate metabolism. Metabolic action of GH occurs in multiple tissues including the liver, muscle, fat and pancreas either directly or indirectly through insulin-like growth factor 1. The diabetogenic action of GH has been well-described in previous in vivo studies. In this paper, we review the metabolic effects of GH on peripheral tissues focusing on glucose metabolism and insulin resistance, and discuss results from human studies on the long-term effects of GH administration on insulin resistance and hyperglycemia.
Introduction
Glucose balance in circulation is tightly maintained within normal range by dynamic regulation of both glucose production (from liver and kidney) and glucose usage by peripheral tissues including the liver, muscle, fat, and kidney [1]. Insulin, the primary regulator of glucose balance, lowers postprandial plasma glucose by increasing glucose uptake and usage from peripheral tissues and decreasing gluconeogenesis (synthesis of glucose from noncarbohydrate precursors such as lactate and alanine) and glycogenolysis (breakdown of glycogen to glucose). In contrast, counterregulatory hormones against insulin action, such as glucagon and epinephrine, prevent hypoglycemia by increasing gluconeogenesis and glycogenolysis and decreasing glucose uptake and consumption from peripheral tissues during fasting. Growth hormone (GH) has been established as one of the counterregulatory hormones, ever since Houssay et al. had found that hypersensitivity to hypoglycemic effect of insulin was increased in hypophysectomized animals and insulin sensitivity was reduced after administration of anterior-pituitary extracts [2]. The diabetogenic effect of GH was also supported by the high prevalence of diabetes in acromegaly patients [1]. The effects of GH on systemic glycemic control is complex partly due to its indirect effects via insulin-like growth factor 1 (IGF-1), which has glucose-lowering effects similar to insulin. For instance, adults with GH deficiency are paradoxically associated with abdominal obesity and insulin resistance, which may be partly associated with their reduced IGF-1 action. This review will summarize the direct and indirect effects of GH on glucose metabolism in peripheral tissues at the molecular level, and also discuss results from human studies investigating the effect of GH administration on glycemic control and insulin resistance.
Effects of GH on glucose metabolism
Studies investigating the effects of GH on glucose metabolism have demonstrated that GH increases glucose production through gluconeogenesis and glycogenolysis from the liver and kidney. Patients with acromegaly and human individuals exposed to high doses of GH showed markedly increased gluconeogenesis activity in the liver and kidney [3]. Additionally, a recent in vivo study demonstrated that GH treatment increased the mRNA expression of 2 major gluconeogenic genes, phosphoenolpyruvate carboxy-kinase and glucose-6-phosphatase, in mouse hepatocytes [4]. GH administration was found to increase glycogenolysis in healthy adults, and inversely, pituitary microsurgery in patients with acromegaly decreased glycogenolysis [5]. Despite the increased glycogenolysis, significantly increased hepatic glycogen contents were reported in transgenic rats overexpressing the human GH gene, suggesting increased glycogen synthesis by excessive GH [6]. Enhanced glycogen synthase activity was also reported in those human GH transgenic rats [6].
Previous studies have shown that GH suppresses glucose uptake in the adipose tissue. An in vivo study demonstrated that GH administration in rats suppresses the amount of glucose transporter 1 (GLUT1) and GLUT4 in adipocyte plasma membrane [7]. Recently, the molecular mechanism of the inhibitory effect of GH on GLUT translocation has been revealed. The insulin-dependent cellular response, which includes trafficking of GLUT4 to the plasma membrane, requires the activation of phosphoinositide 3-kinase (PI3K), a key mediator of metabolic signaling downstream of the insulin receptor. PI3K signaling is negatively regulated by the p85 regulatory subunit. GH was found to induce up-regulation of p85 in white adipose tissues in mice with excess GH production, and in adipocytes treated with GH in an in vitro study [8]. These results imply a mechanism involving GH-induced insulin resistance through up-regulation of the p85 regulatory subunit of PI3K.
GH stimulates lipolysis via activation of the hormone-sensitive lipase, primarily in the visceral adipose tissue, which results in free fatty acid (FFA) flux from adipose tissue to circulation [1]. Previous studies have shown that increased FFA in circulation can induce insulin resistance by inhibition of insulin receptor substrate-1 (IRS-1) activity and subsequent failure of PI3K activation in the skeletal muscle and liver [1]. Meanwhile, increase in FFA uptake by hepatocytes result in promotion of hepatic lipid oxidation and accumulation of acetyl Coenzyme A (Acetyl-CoA). Acetyl-CoA stimulates two key enzymes for gluconeogenesis (pyruvate carboxylase and phosphoenolpyruvate carboxykinase) and an enzyme liberates glucose-6-phosphate as glucose from the liver and kidney into circulation (glucose-6-phosphatase), resulting in an increase of blood glucose levels [9].
In contrast to the GH effects on adipose tissue, GH promotes cellular uptake of FFA in skeletal muscle by increasing the activity of lipoprotein lipase [10]. The re-esterification of triglycerides from FFA results in the accumulation of lipid intermediates such as diacylglycerol and ceramides in skeletal muscle [11]. Previous studies have revealed that diacylglycerol and ceramide impede insulin signaling pathways. Diacylglycerol activates protein kinase C theta, which inhibit IRS-1 through serine phosphorylation, and ceramide inhibits Akt/protein kinase B, an important mediator of the insulin signaling pathway [11]. As in the adipose tissue, up-regulation of the p85 regulatory subunit in skeletal muscle by GH was involved in insulin resistance in mice with excess GH production [12].
The cross-talk between insulin and GH downstream of receptor activation in the skeletal muscle and adipose tissue provides another alternative potent mechanism mediating GH-induced insulin resistance, which is supported by experiments both in vitro and in animal models. IGF-1 production and somatic growth by GH is mediated via the Janus kinase 2 (JAK2)/signal transducer and activator of transcription 5 (STAT5) signaling pathway. GH-induced STAT5 activation increases expression of suppressor of cytokine signaling (SOCS), which interferes with JAK2/STAT5 and consequently downregulates GH action [13]. Crosstalk between GH and insulin receptors occurs at the level of SOCS proteins. Overexpression of SOCS proteins was reported to induce insulin resistance either via inhibition of insulin-induced IRS-1 phosphorylation or via degradation of IRS-1 [14]. Despite numerous in vitro studies and animal models supporting this hypothesis, human studies have failed to demonstrate direct inhibitory effects of GH on insulin signaling pathways in skeletal muscle or fat [15].
Hyperinsulinism after GH administration or in excess GH conditions have been explained by beta-cell compensation for insulin resistance; however, a recent study found that GH directly promotes beta-cell proliferation and glucose-stimulated insulin secretion [16]. In theory, persistently high FFA in chronic excess GH (e.g., acromegaly) may cause beta-cell apoptosis and a subsequent decrease in insulin secretion [17], but in vivo evidence in unclear.
Effects of IGF-1 on glucose metabolism
Excess GH or GH administration in vivo are followed by an increase in circulating IGF-1 levels, therefore net effects of GH on glucose metabolism are complicated by IGF-1 effects. The receptors for IGF-1 and insulin are highly homologous in structure and biological function. Binding of the ligands results in autophosphorylation of the intracellular kinase domains and subsequent activation of downstream signaling cascades, which regulate gene transcription involved in substrate metabolism, cell growth and differentiation. IGF-1 and insulin strongly activate their respective receptors, but can also bind and weakly activate each other's receptors [18]. IGF-1 had caused a hypoglycemic effect in previous in vivo studies through stimulation of glucose uptake and gluconeogenesis, possibly either via activation of IGF-1 or via insulin receptors [19]. For example, administration of recombinant human IGF-1 to insulin receptor deficient mice induced insulin-mimetic effects, including both an increase in glucose uptake of skeletal muscle and a decrease in hepatic gluconeogenesis, through IGF-1 receptor activation. Consistent with this, IGF-1 infusion improved serum glucose levels in individuals with diabetes mellitus [20].
GH treatment and glucose metabolism in GH deficient adults
The clinical presentations of GH-deficient adults are characterized by increased visceral adiposity, insulin resistance, dyslipidemia and hyperglycemia, which contributes to increased risk of cardiovascular morbidity and mortality [2]. Because IGF-1 has anti-inflammatory properties and is important for glucose uptake from peripheral tissues, metabolic disturbances in GH-deficient adults can be explained by the IGF-1 deficit [1]. A deprivation of GH-induced lipolysis and subsequent increased visceral adiposity are also involved in increased circulating FFAs and insulin resistance in these patients [2]. Most of the metabolic disturbances, including visceral adiposity, sarcopenia, hypertension, and dyslipidemia were reported to be relieved after GH treatment [1]. However, a number of studies suggested that there are possible negative impacts on glucose homeostasis such as impaired glucose tolerance as well as insulin sensitivity in patients with GH deficiency after GH administration. Notably, the interpretation of human studies regarding GH treatment and associated changes of glucose metabolism is intricate, because dosage and duration of GH as well as age, body mass index (BMI), and family history of diabetes in study participants can influence the study results.
The original dosage of GH treatment used in GH deficient adults were body weight-adjusted high dosing derived from the dosage used in GH deficient children, however this practice has changed to individualized dosing with lower doses to avoid adverse events of overtreatment since early 2000s. Many of the early studies using high GH doses (≥0.01 mg/kg/day) reported that fasting glucose and insulin levels increased after short-term GH treatment for less than 6 months, but were usually restored to baseline levels after 1 or 2 years of GH treatment (Table 1). GH treatment in high doses was effective for the reduction of total and visceral fat mass [21,22]. However, long-term GH replacement in high doses decreased insulin sensitivity and aggravated insulin resistance, which can be explained by the anti-insulin effects of GH. Despite increased insulin resistance, hemoglobin A1c (HbA1c) levels remained unchanged in both short-term and long-term treatment (Table 1).

Effects of recombinant human GH treatment on glucose metabolism in adults with GH deficiency
Low-dose GH administration in GH-deficient adults has been reported to be effective in improving body composition, albeit to a lesser degree than high-dose GH [23,24]. Most of the studies using low-dose GH treatment (<0.01 mg/kg/day) demonstrated no significant change or just a transient increase in fasting glucose levels (Table 1). Most of these studies reported unchanged insulin resistance and insulin sensitivity after long-term treatment with low-dose GH. Two studies conducted by the same investigators [25,26] showed an improvement of insulin sensitivity in GH-deficient patients with obesity after a short-term treatment with a fixed low-dose GH (0.1 mg/day). Most studies with low-dose GH treatment reported no significant changes in HbA1c levels, although a few studies showed a mild decrease in HbA1c within normal range in GH deficient adults [24]. One study investigated the effect of GH on fasting glucose levels and HbA1c in GH-deficient patients with pre-existing diabetes mellitus, and it revealed a mild elevation of fasting glucose without statistical significance and no aggravation of HbA1c values [23].
Recent studies assessed the risk of development of diabetes mellitus after low-dose GH treatment (Table 2). Because GH-deficient adults are at increased risk of impaired glucose tolerance compared to the general population, it is sufficient to compare the incidence of diabetes in GH treated patients with that of untreated patients. Currently, only one study with relatively short duration (2.3 years) reported that there was no increased risk of diabetes mellitus in GH-deficient adults after GH treatment compared with untreated GH-deficient controls [27]. Other studies compared the incidence of diabetes between GH treated patients and the normal general population, and most of them revealed no significant increase in the incidence of diabetes after 2–10 years of GH treatment (Table 2). One study demonstrated 2–6 times higher incidence of diabetes mellitus than expected in the normal general population [28]. It is noteworthy that increased age and BMI, female sex, and duration of GH, but not the dose of GH, were associated with increased risk of diabetes mellitus in this study [28].

Effects of recombinant human GH treatment on the development of DM in adults
GH treatment and glucose metabolism in GH deficient children and adolescents
To date, relatively few studies have been conducted examining the change of glucose metabolism after GH treatment in the pediatric population. Most of these studies have demonstrated that increased insulin resistance, indicated by increased fasting insulin and homeostasis model assessment of insulin resistance levels, was observed during GH therapy in GH-deficient children and adolescents, but their fasting/postprandial glucose and HbA1c levels remained withinin normal range (Table 3). A limited number of euglycemic clamp studies reported decreased insulin sensitivity in GH-deficient children after short-term GH treatment [29,30].
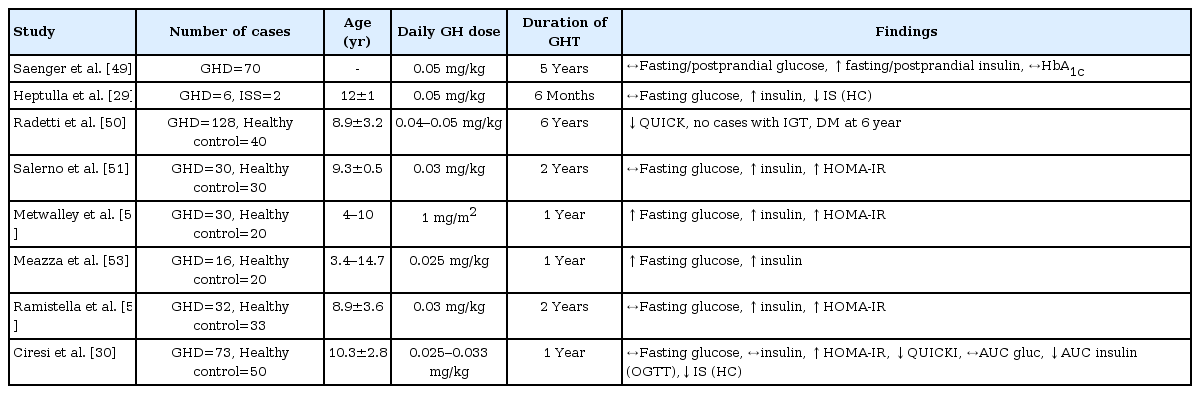
Effects of recombinant human GH treatment on glucose metabolism in children and adolescents with GH deficiency
Because increased insulin resistance and impaired insulin sensitivity are linked to the risk of glucose intolerance and diabetes mellitus, concerns have been raised regarding the possible development of diabetes mellitus during or after GH treatment in the long-term (Table 4). With the advent of National Cooperative Growth Study research [31], large pharmacoepidemiological studies have demonstrated that the incidences of type 2 diabetes mellitus increased more than 6 times in children under GH treatment compared with the general population, especially in patients with predisposing risk factors for diabetes, such as obesity, family history of diabetes, Turner syndrome, Prader-Will syndrome, or glucocorticoid treatment [32,33]. The development of diabetes mellitus was not associated with the dose or duration of GH treatment, and the incidence of type 1 diabetes mellitus during GH treatment was comparable with that of the general population in all three studies [31-33]. In comparison, a recent French population-based study reported that the prevalence of diabetes mellitus in GH-treated children was similar to the general population when patients reached early adulthood [34]. Of note, this study included patients with isolated GH deficiency, idiopathic short stature, or short children born small-for-gestational age, excluding patients with high risk of mortality and morbidity, such as patients with cancer, chronic renal failure, multiple pituitary hormone deficiency, Turner syndrome or Prader-Willi syndrome [34].

Effects of recombinant human GH treatment on the development of DM in children and adolescents
Summary
GH therapy antagonizes insulin's action on peripheral tissues, such as the skeletal muscle, liver, and adipose tissue, thereby increases glucose production from the skeletal muscle and liver and decreases glucose uptake from adipose tissue. Insulin production is increased to compensate the increased circulating glucose after GH administration. GH-induced lipolysis in the visceral adipose tissue and subsequent increased circulating FFA also interferes with insulin signaling pathways, and chronic exposure to high FFA may exert direct toxicity in beta-cells. Meanwhile, IGF-1 has insulin-mimetic actions in the skeletal muscle and liver, and increased circulating IGF-1 after GH administration may have beneficial effects on glucose homeostasis and insulin resistance.
A number of human studies have suggested that GH administration in GH deficient adults may reduce visceral adiposity and improve cardio-metabolic disturbance. However, some studies raised concerns over increased insulin resistance and impaired fasting glucose during GH treatment, especially in patients with obesity and elderly patients. Studies in children and adolescents also suggested that GH administration may induce insulin resistance in short-term treatment, but its long-term consequences have not been fully determined yet. International cohort studies indicate that GH therapy may increase the incidence of type 2 diabetes mellitus in children and adolescents with predisposing risk factors, therefore it is prudent to monitor possible negative consequences on glucose metabolism during and after GH administration. Large-scale longitudinal cohort studies are required to examine the long-term influence of GH therapy on cardiovascular outcomes in GH-deficient children with or without continuation of GH after cessation of skeletal growth.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.