Introduction
Hemochromatosis is an inherited or acquired disorder caused by excessive iron storage1). Primary hemochromatosis is an autosomal recessive disease caused by gene mutations related to iron absorption, and it presents as liver cirrhosis, cardiac dysfunction, hypothyroidism, diabetes, and hypogonadism. Secondary hemochromatosis usually develops in patients who have received multiple red blood cell transfusions2). Recurrent transfusion is a rapid and effective treatment for refractory anemia, including severe aplastic anemia, myelodysplastic syndrome, and thalassemia; however, transfusion-associated iron overload can develop as a complication. Excessive iron is deposited in reticuloendothelial cells and parenchymal tissues, and its accumulation can cause progressive damage in multiple organs3).
Endocrinopathies such as hypogonadotropic hypogonadism and osteoporosis are commonly associated with transfusion-related hemochromatosis, and diabetes is the most frequently encountered problem4). Insulin resistance and reduced early insulin secretion caused by iron deposition to the pancreatic islets have been implicated in the main pathophysiological mechanism that leads to diabetes in patients with secondary hemochromatosis. Herein, we describe 2 cases of diabetes mellitus in patients with severe aplastic anemia following multiple transfusions.
Case reports
Case 1
Subject 1, a 15-year-old man, was diagnosed as having severe aplastic anemia, and he was treated with antithymocyte globulin and cyclosporine. He also received multiple red blood cell transfusions (a total volume of 223 units over 7 years). An iron-chelating agent, deferoxamine (1,000 mg/day) was administered because of an increased serum ferritin level of 2,842.6 ng/mL after 1 year of the diagnosis, but his serum ferritin level was persistently high at regular follow-up visits.
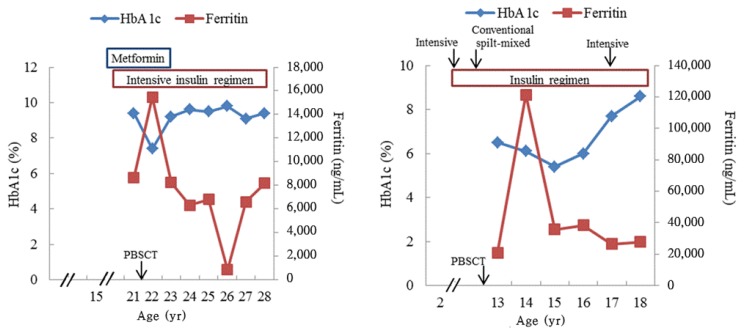
At 21 years of age, hyperglycemia (serum glucose level, 358 mg/dL) was detected on routine laboratory tests with a high body mass index (36.5 kg/m2), and the glycated hemoglobin A1c level was 9.4%. The 24-hour urine C-peptide level was 24.8 ┬Ąg/day (normal range, 23ŌĆō148 ┬Ąg/day) with negative antiglutamic acid decarboxylase (anti-GAD) antibody. He had no family history of diabetes or pancreatic diseases, and he did not take any medicine that interfered with glucose metabolism at that time. Results of the serum biochemical test indicated an aspartate aminotransferase level of 94 IU/L, alanine aminotransferase level of 157 IU/L, alkaline phosphatase level of 74 IU/L, and total bilirubin level of 1.7 mg/dL. The serum ferritin and iron levels were 11,106 ng/mL (normal range, 20ŌĆō320 ng/mL) and 270 ┬Ąg/dL (normal range, 65ŌĆō157 ┬Ąg/dL), respectively. The total iron binding capacity was 288 ┬Ąg/dL (normal range, 256ŌĆō426 ┬Ąg/dL). An echocardiogram showed a normal ventricular chamber size and good ventricular function, indicating no evidence of cardiac involvement of hemochromatosis. However, an abdominal computed tomography scan showed hepatosplenomegaly and nonspecific, multiple small lymph node enlargement in the celiac and periportal regions. Subsequently, metformin was administered; however, his glucose level was poorly controlled, with the glycated hemoglobin A1c level increasing as high as 9%, which required an intensive insulin regimen (Fig. 1A).
At 22 years of age, he underwent the second allogeneic peripheral blood stem cell transplantation (PBSCT) after 3 months of the first allogenic PBSCT from his mother because of graft failure. During the follow-up period, mild sensory and motor neuropathies were observed at 24 years of age. Currently, he is 28 years old and has shown no evidence of cardiovascular complications with a normal blood pressure and low-density lipoprotein cholesterol level. Although his peripheral neuropathy did not progress, his urine albumin/creatinine ratio has gradually increased to 582.5 mg/g causing him to require angiotensin-converting-enzyme inhibitor medication. Results of endocrine tests were normal: thyroid-stimulating hormone (TSH) level of 3.2 ┬ĄU/mL, free thyroxine (T4) level of 1.5 ng/dL, luteinizing hormone (LH) level of 2.3 mIU/mL, follicle-stimulating hormone (FSH) level of 3.2 mIU/mL, testosterone level of 2.5 ng/mL, and insulin-like growth factor-I (IGF-I) level of 164 ng/mL (Table 1). His height and weight were 181.1 cm (1.36 standard deviation score [SDS]) and 96 kg (2.29 SDS), respectively. The sexual maturity rating of the testis and pubic hair was Tanner stage V, and the testicular volume of both of his testes was 8 mL. As his physical sign of puberty progress, dynamic studies evaluating hypothalamic-pituitary-gonadal axis were not performed.
Case 2
Subject 2 was diagnosed as having severe aplastic anemia at 2 years of age, and she received multiple red blood cell transfusions before she underwent haploidentical PBSCT. Diabetes mellitus developed at 11 years of age with a high serum ferritin level (12,559.8 ng/mL). An intensive insulin regimen was initiated, and her blood glucose level was well controlled; her hemoglobin A1c level was less than 7% until she was referred to our pediatric hemato-oncology clinic at 13 years of age. Laboratory data obtained before she was referred were unavailable. At the time of admission, her skin was relatively dark with pale conjunctiva, and the examination of the lungs, heart, and abdomen was unremarkable. Peripheral blood test results showed a white blood cell count of 1,500/mm3 (absolute neutrophil count, 150/mm3), hemoglobin level of 10.1 g/dL, and platelet count of 39,000/mm3. The glycated hemoglobin A1c level was 6.6%, and the 24-hour urine C-peptide level was 107 ┬Ąg/day with a negative anti-GAD antibody. The amount of transfused red blood cells could not be estimated because of inadequate data from the previous hospital. Liver and renal function was normal. At 13 years of age, she underwent haploidentical PBSCT from her mother after a conditioning regimen composed of antilymphocyte globulin, cyclophosphamide, and fludarabine with total body irradiation was administered. At 14 years of age, endocrine test results showed the following: TSH level of 0.96 ┬ĄU/mL, free T4 level of 1.1 ng/dL, LH level of 2.5 mIU/mL, FSH level of 2.5 mIU/mL, estradiol level of 16.4 pg/mL, cortisol level of 2.5 ┬Ąg/dL, and IGF-I level of 200 ng/mL (Table 1). On physical examination, height and weight were 150.5 cm (ŌłÆ1.57 SDS) and 45.8 kg (ŌłÆ0.81 SDS), respectively. The sexual maturity rating of breast and pubic hair was Tanner stage II. A combined anterior pituitary hormone test was performed at 14 years of age because of her short stature and delayed puberty. Peak levels of LH and the growth hormone (GH) were 3.1 mIU/mL and 0.79 ng/mL, respectively. Other pituitary hormone responses were normal. Her bone age was 13 years according to the Greulich and Pyle method5). Magnetic resonance imaging of the pituitary gland was not performed. However, the combination of hypogonadotropic hypogonadism and GH deficiency indicated damage in the pituitary gland, and both total body irradiation and iron accumulation could be considered as causes of pituitary dysfunction. After confirming the diagnosis, estropipate (0.16 mg/day, Esgen) was started, and the dose was gradually increased to 0.94 mg/day.
Additionally, bone mineral densitometry results showed osteoporosis, with a z-score of ŌłÆ3.7 in the femur and ŌłÆ2.8 in the spine, thus calcium carbonate/cholecalciferol (1,500 mg/day, Cal-D-vita, Roche). Supplementation therapy was also initiated. As iron overload caused organ dysfunction, we performed phlebotomy 4 times instead of administering iron-chelating agents, and the serum ferritin level gradually decreased to 2,763.2 ng/mL. Her blood glucose level was initially well controlled, and the glycated hemoglobin A1c level remained lower than 7% with a conventional split-mixed insulin regimen until 6 years after the diagnosis of diabetes (Fig. 1B). However, frequent blood glucose excursions and gradually increasing levels of glycated hemoglobin A1c were observed, so multiple daily insulin injections were initiated to control hyperglycemia at 17 years of age. She is currently 18 years old, and she has been receiving treatment with multiple insulin injections and fenofibrate (160 mg/day) for hypertriglyceridemia, and estrogen/progesterone replacement therapy. Microvascular complications were not observed.
Discussion
This study reports 2 cases of diabetes mellitus caused by secondary hemochromatosis in pediatric age. Endocrine dysfunction in transfusion-associated hemochromatosis has been reported in adults4). The present study described clinical presentation and long-term course of adolescent patients with endocrinopathy caused by transfusion-associated hemochromatosis.
The loss of insulin secretory capacity and insulin resistance plays a role in the pathogenesis of diabetes mellitus in hemochromatosis6). Recently, insulin resistance and impaired insulin secretion have been suggested as the main pathophysiological mechanism that leads to diabetes by hemochromatosis7). Thus, diabetes mellitus due to hemochromatosis may primarily develop because of impaired insulin secretion caused by ╬▓-cell dysfunction, and it is accelerated by variable factors affecting insulin resistance, e.g., cirrhosis or obesity.
Severe aplastic anemia is a potentially life-threatening disease involving bone marrow failure, which is characterized by pancytopenia. Many patients with severe aplastic anemia require repeat red blood cell transfusions as conservative therapy, although they are mainly treated with immunosuppressive agents or hematopoietic stem cell transplant8). Therefore, these multiple transfused patients are at an increased risk of iron overload and its related complications9).
Hemochromatosis develops from excessive iron absorption or impaired iron recycling1). The human body normally maintains an iron level at 3 to 4 g by absorbing the same amounts of iron and regular daily loss of iron: 1.5 mg for women and 1 mg for men10). However, patients with hemochromatosis absorb 4 mg of iron per day, which increases blood iron levels, resulting in increased blood levels of ferritin and the accumulation of iron in multiple organs. Repeat transfusion is one of the secondary causes of hemochromatosis. One unit of red blood cells contains 200ŌĆō250 mg of iron; therefore, iron overload can occur after the transfusion of more than 20 units of red blood cells because of the body's limited capacity to excrete excess iron. Excessive iron is stored in reticuloendothelial cells when the iron-capturing ability of macrophages reaches its limit, and it is impregnated into multiple organs such as the heart, liver, spleen, bone marrow, and endocrine system, resulting in organ failure11). It may also reduce the function of erythroid progenitor cells by affecting their proliferation.
Patients with hemochromatosis may present with heart failure, hepatomegaly, liver dysfunction, hypogonadism, diabetes mellitus, or arthropathy. Endocrinopathies are common complications in patients with transfusion-associated hemochromatosis12), and the most frequently encountered complication is diabetes mellitus. Diabetes mellitus is especially a significant complication of hemochromatosis; therefore, numerous studies have been conducted to demonstrate a positive relationship between diabetes and iron stores. In a meta-analysis of 4,366 participants with type 2 diabetes, the relative risks, when comparing the highest and lowest category of ferritin levels, were 1.66 (95% confidence interval [CI], 1.15ŌĆō2.39) for prospective studies and 2.29 (95% CI, 1.48ŌĆō3.54) for cross-sectional studies13). Another recent prospective cohort study described the relationship between the ferritin concentration and type 2 diabetes in 1,613 Finnish men14). Three hundred thirty-one patients with incident diabetes had a significantly higher ferritin level than those who remained healthy (191 ┬Ąg/L vs. 151 ┬Ąg/L, PŌēż0.001).
Therapeutic phlebotomy is the only broadly accepted treatment for hemochromatosis. In previous studies, phlebotomy improved insulin secretory function without any significant change in insulin sensitivity, but it impaired glucose metabolism in patients with hemochromatosis15,16). It has been suggested that the early initiation of phlebotomy can improve insulin secretion and stop disease progression, although damage to ╬▓-cells and the insulin secretory defect may be irreversible in those with advanced disease. The worldwide consensus recommends maintaining the iron level between 50 and 100 ┬Ąg/L with regular monitoring17,18). As an alternative therapy, iron-chelating agents have also been used in patients with hemochromatosis, because they eliminate excessive iron effectively and induce the rapid restoration of normal ferritin concentrations19,20).
In conclusion, patients with secondary hemochromatosis due to repeat red blood cell transfusions may develop endocrine disorders, among which diabetes is the most common problem caused by insulin resistance and impaired insulin secretion. Thus, it is crucial to limit transfusions and conduct regular follow-up visits for patients with hemochromatosis.