Introduction
Noonan syndrome (NS) is an autosomal dominant disorder with an estimated incidence of 1:1,000 to 1:2,500 live births1). It is characterized by short stature, distinctive facial appearance, congenital heart defects (most frequently pulmonary valve stenosis or hypertrophic cardiomyopathy), thoracic deformities, bleeding diathesis, and cryptorchidism2).
Tartaglia et al.3) demonstrated that the causative gene of NS is PTPN11 on chromosome 12q24.1, encoding the protein tyrosine phosphatase, SHP-2, in 40% to 50% of cases. The other common causative genes of NS are SOS1 (17%–28%) and RAF1 (5%–17%)4,5,6,7,8). Genes accounting for less than 5% of NS are KRAS, NRAS, and BRAF8,9). Other rare causes of NS are MEK1 and RIT1 mutations9,10). Mutations in the SHOC2 and CBL genes were reported in Noonan-like syndrome with loose anagen hair (NS/LAH)7,11).
NS is a genetically heterogeneous disorder caused by up-regulated RAS-MAPK signaling, resulting in growth disturbances4). The RAS-MAPK cascade is activated in response to cytokines, hormones, and growth factors, and is a major mediator of early and late developmental processes, including morphology determination, organogenesis, synaptic plasticity processes, and growth5). SHP-2, a protein tyrosine kinase encoded by PTPN11, plays diverse roles in signal transduction via the RAS-MAPK pathway, such as growth hormone (GH) receptor signaling in children with NS12). Thus, short stature with delayed bone age is the most common clinical feature of NS13), with a mean adult height below –2 SDS14). Over the last 2 decades, recombinant human GH (rhGH) treatment has been reported to increase height SDS in NS patients without severe adverse events15). Long-term rhGH therapy for about 4.2–11.8 years in NS has been reported to produce height gains varying from 0.6 to 2.0 SDS16,17).
Our group previously reported that 1 year of rhGH therapy significantly improved height SDS in NS patients and the response to rhGH therapy was not affected by PTPN11 mutations18). However, the long-term efficacy and safety of rhGH therapy in NS patients has remained elusive. Thus, this study evaluated the effects of long-term treatment with rhGH in NS patients and the influence of mutations in RAS-MAPK pathway genes.
Materials and methods
1. Subjects
This study included 11 males and 4 females with NS who received rhGH therapy for at least 3 years. Mean duration of rhGH therapy was 4.9 years (range, 3.3–6.6 years). The diagnosis of NS was based on the van der Burgt criteria19). Baseline characteristics of the 15 subjects are shown in Table 1. Among the 15 children, 12 had congenital heart defects, including pulmonic stenosis (7 subjects, 46.7%), ventricular septal defect (3 subjects, 20%), patent ductus arteriosus (4 subjects, 26.7%), atrial septal defect (2 subjects, 13.3%), or right ventricular defect (1 subject, 6.7%). Five of them had two or more congenital heart defects. Mutations in the PTPN11 gene were identified in 9 subjects (60%). Mutations in SOS1 were identified in 2 children (13.3%) and KRAS mutation in 1 child (6.7%) (Table 1). No mutations were identified in 3 of the subjects (20%).
2. Methods
The rhGH (Norditropin; Novo Nordisk Pharma, Hellerup, Denmark) was administered at a dose of 50–75 µg/kg/day for 6 days a week subcutaneously. The main outcome measures were height SDS, growth velocity (GV), bone age, serum insulin-like growth factor 1 (IGF-1) SDS, and IGF binding protein 3 (IGFBP-3) SDS. The subjects' height and weight were measured at baseline and every 6 months. Height and weight were expressed as SDS based on normative data from Korean references20). The complete blood count, routine chemistry, free T4, thyroid-stimulating hormone, IGF-1, and IGFBP-3 levels were measured at baseline and at 6-month intervals. IGF-1 SDS and IGFBP-3 SDS were calculated based on normative data from the Korean reference21). The subjects' bone age was determined annually using the Greulich-Pyle method22). Electrocardiogram and echocardiogram were carried out every 6 months.
Puregene DNA isolation kits (Gentra, Minneapolis, MN, USA) were used for genomic DNA extraction from peripheral blood leukocytes. The coding regions and intronic flanking regions of the PTPN11 (whole exons), SOS1 (whole exons), KRAS (exons 1 and 4), RAF1 (exons 7, 14, and 17), SHOC2 (exon 1), NRAS (exon 3), BRAF (exons 6, 11, and 16), and MEK1 (exon 3) genes were amplified by polymerase chain reaction with specific primers and directly sequenced using an ABI3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA).
3. Statistical analysis
The Friedman test was used to evaluate the effect of rhGH therapy. The Wilcoxon signed rank test was used to compare the changes between pre- and post-rhGH therapy. The relationships between genotypes of subjects and growth parameters, such as bone age, height SDS, GV, and serum IGF-1 and IGFBP-3 levels were assessed by the Mann-Whitney U-test. Statistical analyses were conducted using IBM SPSS Statistics ver. 21.0 (IBM Co., Armonk, NY, USA). P<0.05 was considered statistically significant.
Results
1. Efficacy of rhGH therapy in children with NS
The mean age at the start of treatment was 7.97±1.81 years (range, 5.7 to 11.3 years). Height SDS, GV, and serum IGF-1 SDS levels were significantly increased after rhGH therapy (Table 2). Height SDS increased from –2.64±0.64, to –1.54±1.24, to –2.13±1.08, respectively, at the first, second, and third years of treatment (P=0.005, P=0.003, and P=0.001, respectively). GV during the first year of treatment was highest (8.57±1.49 cm/yr). GV increased from 4.64±0.80 cm/yr at baseline to 6.79±1.26 and 6.41±1.54 cm/yr at the second and third years (P=0.001 and P=0.003, respectively). Serum IGF-1 SDS significantly increased from –1.28±1.03 to –0.10±0.94 after 3 years (P<0.001). Serum IGFBP-3 SDS changed from –0.15±0.40 to –0.09±0.34, which was not statistically significant (P=0.074). Bone age increased from 5.09±2.12 years to 9.42±2.15 years after 3 years (P<0.001). The bone age/chronologic age ratio increased from 0.62±0.13 at the start of treatment to 0.68±0.15, 0.79±0.1, and 0.86±0.1, respectively, after 1, 2, and 3 years of treatment and the end of treatment (P=0.02, P=0.001, and P=0.001, respectively). During treatment, there were no serious adverse events including the cardiac dysfunction, hypertrophic cardiomyopathy, malignancy, hyperglycemia, or thrombocytopenia with bleeding tendency.
2. Response to rhGH therapy according to genotypes
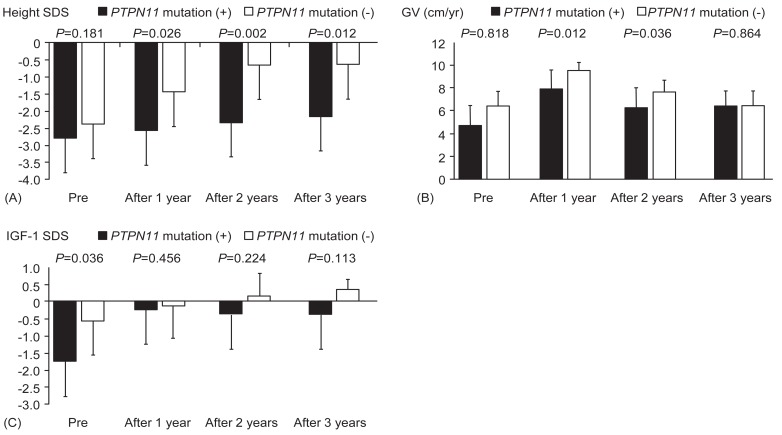
The influence of genotype was analyzed by comparing the growth parameters at the start of treatment and 1 year, 2 years, and 3 years after treatment. IGF-1 SDS was significantly different between the group with PTPN11 mutations and the group without PTPN11 mutations at the start of treatment (P=0.036). The other baseline data, including bone age, height SDS, GV, and serum IGFBP-3 levels, were not significantly different between the 2 groups (P=0.607, P=0.181, P=0.818, and P=0.224, respectively) (Fig. 1). Responses to treatment over 3 years, represented by changes in bone age, GV, serum IGF-1 SDS, and IGFBP-3 SDS, were not significantly different among children with and without mutations in PTPN11 (P=0.755, P=0.864, P=0.113, and P=0.145, respectively). However, height SDS was significantly increased in patients without PTPN11 mutations compared to those with mutations (P=0.012) (Fig. 1).
Discussion
This paper demonstrated a significant increase in growth parameters, including bone age, height SDS, and IGF-1 SDS in children with NS after 3 years of rhGH therapy. In a previous study with 30 subjects, rhGH therapy increased GV by 2 cm/yr or more in 80% of the children after 12 months of rhGH therapy23). Long-term rhGH therapy for 3 years significantly increased height SDS from –2.7±0.40 to –1.9±0.9 in 23 NS children (P<0.001) compared to 8 untreated patients24). However, GV acceleration was not significant during the second and third years (P=0.4 and P=0.5, respectively)24).
Several studies have reported the association between PTPN11 mutation and GH resistance by a postreceptor signaling defect25,26). The GH resistance in NS children with PTPN11 mutations may contribute to short stature and their relatively poor response to rhGH25). Mean GH levels showed a tendency to be higher in the PTPN11 mutation-positive group (P=0.075)25). However, IGF-1 and IGFBP-3 SDS levels were significantly lower in the PTPN11 mutation-positive group than in the PTPN11 mutation-negative group (P=0.006)25). The improvement of height SDS after 1 year of rhGH therapy was significantly lower in the PTPN11 mutation-positive group (P=0.007)25). In a prospective multicenter study in 35 NS patients, rhGH therapy for 2 years resulted in increased height SDS, which was significantly lower in the PTPN11 mutationpositive group than the PTPN11 mutation-negative group (P=0.03)26). Those findings were consistent with a recent study with an animal model showing reduced sensitivity to GH in PTPN11-mutated mice27). In contrast, the response of height SDS to GH treatment for 3.0–10.3 years was not significantly different between patients with or without PTPN11 mutations (P=0.98)16). In the present study, there was a significant difference in height SDS between children with and without PTPN11 mutations after 1, 2 and 3 years of rhGH therapy. GV was significantly different between 2 groups after 1 and 2 years of rhGH therapy. However, GV at third year was not significantly different between 2 groups, suggesting growth response was blunted after long-term rhGH therapy.
Cardiac anomaly is one of the most important characteristics of NS2). As pulmonary valve stenosis (30%–39%) and hypertrophic cardiomyopathy (9.5%–30%) are frequent problems in NS patients, careful monitoring is recommended to detect hypertrophic cardiomyopathy and progression of the underlying heart disease during the period of rhGH treatment28,29). From prospective rhGH trials over 3 years, no children with NS experienced any heart problems based on echocardiography17,30). Two patients have been reported to have mild progression of pulmonary valve stenosis, which was considered to be unrelated to rhGH therapy16). There were no cardiac complications during rhGH therapy in the current study.
In conclusion, long-term rhGH therapy in NS patients was safe and effective at improving height SDS, GV, and serum IGF-1 levels in accordance with previous studies. However, the meticulous monitoring of potential adverse events is still needed because of high dose of rhGH and preexisting hyperactivity of RAS-MAPK pathway. Height SDS in patients without PTPN11mutation was significantly increased compared to those with PTPN11 mutation after 3 years of rhGH therapy.