Early onset of puberty in an obese boy with Klinefelter syndrome
Article information
Abstract
Klinefelter syndrome (KS) is one of the most common disease entities characterized by X-chromosomal aberration causing the primary hypogonadism in adult men. Patients with KS seem to be typically characterized by tall, slender bodies with delayed puberty and hypergonadotropic hypogonadism. However, it has been known that they have a broad spectrum of phenotype ranging from almost normal external appearances to typical phenotype. Only 25% KS Patients are ever diagnosed because KS remains unrecognized. Also, boys with KS have an onset of pubertal development within the normal range, not delayed onset of puberty. Adolescents with KS are generally diagnosed as having the lack of pubertal progress. Early detection of KS can be difficult without awareness. We report an unusual case of early onset of puberty in obese boy with KS who presented with a unilateral non-hormone secreting testicular teratoma.
Introduction
Klinefelter syndrome (KS), characterized by the presence of one or more additional X chromosomes, occurs in approximately 1 in 1,000 live male births1). The classical 47, XXY karyotype is presented in about 90% of the KS. Its clinical characteristics are generally known as classic physical features like tall eunuchoidal body proportions, gynecomastia, small testes with micropenis, a lack of virilization, infertility, and hypergonadotropic hypogonadism male due to the primary testicular dysfunction2). But alternative phenotype with fewer patients has now been recognized3) and no exclusive symptom excluding small testes and high gonadotropins levels in adulthood defines the syndrome. So KS frequently remains unrecognized and only 25% of KS Patients are ever diagnosed4).
Also, testes volume is within the normal range in KS children and male hypogonadism seldom occurs in prepubertal KS boys. Boys with KS have an onset of pubertal development within the normal range, not delayed onset of puberty. Decreased awareness of KS results in a delayed, under diagnosed state.
We report an obese boy with early onset of puberty who was diagnosed with KS and unilateral nonhormone secreting testicular tumor.
Case report
A 10.6-year-old boy was referred to the Department of Urology in Inha University Hospital for evaluation of different sizes in both testes. At a Doppler-ultrasound sonography, there were an ill-defined hyperechoic lesion with microcalcification and varicocele in his left testis. He had undergone left orchiectomy for a testicular tumor on the same side confirmed by frozen biopsy specimens. The histopathologic examinations indicated that the resected testicular mass was mature teratoma (0.30×0.3 cm-sized) with severe testicular atrophy and surrounding tissue had a marked decrease in the number of germ cells (Fig. 1). On karyotyping, the patient was diagnosed with a typical type of KS with 47, XXY. Then, he was referred to our clinic for evaluation of his abnormal karyotype.

(A) Resected left testicular mass revealed a mature teratoma. The nodule in the testis showed mature squamous cell nests in the fibrotic stroma (H&E, ×200). (B) The testicular parenchyme showed atrophic seminiferous tubule without germ cells. The hyperplastic Leydig cells were noted (H&E, ×200).
His birth weight was 3.0 kg. There was no notable family history. His father and mother's height was 174 cm and 158 cm. His parent's pubertal developments were within normal range. Although he showed an average level of academic achievements, he was having difficulty forming normal peer relations because of his shy and passive characters. He had grown taller than his peers since early childhood. Since one year before our clinic visit, the patient had begun to markedly increase in stature and body weight with progressive pubertal changes in his external genitalia. Physical examination showed tall stature (158 cm, >97 percentile) with body weight of 64 kg (>97 percentile), increased body mass index (BMI) (25.6 kg/m2, 95th–97th percentile) and his waist circumference of 93 cm (>97th percentile; 50th percentile, 66.9 cm). His pubertal stages were the gonad on the right side of Tanner III (the right testicular volume 8 mL, penile length 8 cm, width 2.5 cm) and the pubic hair of Tanner II.
The findings of initial blood examinations were as follows: hemoglobin, 13.1 g/dL; hematocrit, 40.1%; white blood cell counts, 7,680×103/µL; alkaline phosphatase, 1,415 IU/L; aspartate aminotransferase/alanine aminotransferase, 35/24 IU/L; insulin-like growth factor-I, 441 ng/mL; beta-human chorionic gonadotropin (β-hCG), 0.86 mIU/mL (normal range, 0.00–2.00 mIU/mL); α-fetoprotein, 1.2 mIU/mL (normal range, 0.0–20.0 mIU/mL). The results of luteinizing hormone-releasing hormone stimulation test at postorchiectomy showed remarkable elevation of luteinizing hormone (LH) and follicle stimulating hormone (FSH) levels (Table 1). Sex steroid hormone levels were estradiol, 46.51 pg/mL (normal range, 5.0–16.0 pg/mL), testosterone, 1.35 ng/mL (normal range, 2.67–10.12 ng/mL), and dehydroepiandrosterone-sulfate (DHEA-S), 59.4 µg/dL (normal range, 80.0–560.0 µg/dL). The findings of thyroid function test were normal. Homeostatic model assessment-insulin resistance was 2.36 (fasting glucose, 95 mg/mL; fasting Insulin, 10.10 mIU/L) and glycosylated hemoglobin (HbA1c) level was 6.0% (Table 2). His bone age was 13 years old by the Greulich and Pyle method at a chronological age of 10.6 years. The finding of brain magnetic resonance imaging was normal. On bone mineral density examination, he had a z-score of 1.2 for the L-spine, which was within the expected range for age.
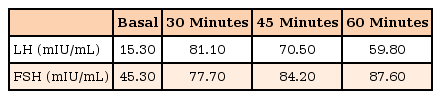
Results of LHRH stimulating test of the patient

Laboratory characteristics of the patient
Since then, he has been undergone regular follow-up to get a psychiatric counselling as well as to monitor the degrees of virilization and obesity without testosterone replacement therapy. On physical examination of the patient aged 13.4 years, his height was 172 cm (90th–95th percentile) and postoperative growth velocity of height is 14 cm/2.6 years. His weight of 82 kg (>97th percentile) and the BMI was 27.7 kg/m2 (95th–97th percentile). His puberty had slow progressed as much as the right gonad of Tanner II–IV and a pubic hair of Tanner III. His bone age was advanced at 16 years old. Follow-up laboratory findings were LH, 7.94 mIU/mL, FSH, 20.8 mIU/mL, estradiol, 36.56 pg/mL, testosterone, 2.47 ng/mL, DHEA-S, 143.3 µg/dL, and HbA1c, 5.8% (Table 2).
Discussion
In general, KS patients are likely to be diagnosed after puberty onset because overt phenotype becomes clear after puberty. Boys with KS have the beginning of puberty within the normal ages. Of boys with delayed onset of puberty, only 5%–10% of them revealed as having hypergonadotropic hypogonadism including KS5). Adolescents with new cases of KS are commonly diagnosed as having the lack of pubertal progress at an average age of 14 years6).
Otherwise our patient was incidentally detected KS early at the age of 10.6 years after surgery for a non–β-HCG secreting teratoma in unilateral testis. He unusually showed obese phenotype with early onset of puberty. The relationship between KS and precocious puberty has been described rarely in the literature. An idiopathic central origin has been reported in a few KS boys7). KS boys are at increased risk of precocious puberty owing to β-HCG secreting tumors7). However, our case had an endocrinologically nonfunctioning tumor. In addition, histopathologic findings in his resected testis showed severe testicular atrophy with marked decrease in testicular germ cell numbers as one of the classic characteristics in KS. Even after the removal of unilateral testis with tumor, a progression of puberty continued for over 2 years in our case. Therefore, because the histopathological findings of his opposite side of testis would be similar to that of the resected testis, his early onset puberty and pubertal progress might be caused by not testicular origin but adrenal origin. Inversely, onset of puberty in KS adolescence may be associated with accelerated germ cell depletion in relation to an increase in testicular volume8).
Most of the male newborns with the 47,XXY karyotype have normal phenotype with external genitalia at birth. On the other hand, germ cell degeneration begins since the midfetal life and progresses to infants and then accelerated during puberty. With the development of fibrosis of seminiferous tubules and hyperplasia of Leydig cells, small firm testes and azoospermia eventually appear. During prepubertal period, inhibin B and anti-müllerian hormone as functional markers of Sertoli cells show normal blood levels and the blood concentrations of testosterone, LH and FSH are also normal. With the onset of puberty, there is a marked increase in serum levels of LH and FSH. This is accompanied by various subnormal serum testosterone concentrations and decreased virilization. In boys with KS, testicular volume peaks at midpuberty, and thereafter testicular growth ceases, simultaneously with development of the hypergonadotropism typical of adult KS patients910).
Our case showed marked increases in serum LH, FSH levels and relative subnormal serum testosterone level. Based on these results, it seems that our case had relative hypergonadotropic hypogonadism in spite of early puberty2111213). So his progress of puberty should cease with development of the hypogonadism during a long-term follow-up period.
The initiation time of testosterone supplementation in KS is determined depending on the onset of hypogonadism. During the early puberty stage of KS patient, testosterone therapy is not normally needed due to usually nondeficient in testosterone4). Selective early initiation of testosterone replacement might be effective for the prevention of gynecomastia and helpful for a decreased body fat mass. During the late-puberty stage, the treatment is initiated after the assessment of testosterone insufficiency. Although a progression of puberty maintained even after unilateral orchiectomy in this case, testosterone supplement therapy is under consideration.
KS Patients have a broad spectrum of phenotype ranging from almost normal external appearances to typical phenotype. These diverse characteristics are closely associated with the length of the GAGn polymorphism of the androgen receptor gene5). A general perception is that most KS patients present with typical phenotype like tall eunuchoidal body shape. This leads to delayed-diagnosis and under-diagnosis which has seen in up to 75% of total KS cases4). Our case also could not be suspected of having KS based on external appearances. He is rather obese and only suffered from a difficulty in forming a relationship with peers because of his passiveness. Recent studies have suggested that such symptoms as learning difficulties, delayed speech, cognitive dysfunctions and difficult peer interactions might be a clue to the early diagnosis of KS14).
Up to 50% of patients with KS tend to have problems with motor and language development during the childhood. They are at increased risk of developing psychiatric disorders, autism spectrum disorders or attention deficit hyperactivity disorder during the adult period. Therefore, boys with neurodevelopmental problems should be suspected of having KS. Also once diagnosed with KS, they are recommended to undergo early psychiatric consult and intervention.
In this report, we described a rare case of early onset of puberty in an obese boy with KS who presented with a unilateral nonhormone secreting testicular tumor. In conclusion, we suggest that even though an onset of male puberty is earlier than normal, when the psychosocial problems are accompanied, KS should be considered.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.