Introduction
Encephalocraniocutaneous lipomatosis (ECCL) is a rare, sporadic neurocutaneous syndrome that was first described as ectomesodermal dysgenesis by Haberland and Perou in 19701). It involves lesions in the eyes, skin, and central nervous system (CNS)2). Ocular lesions typically consist of conjunctival choristoma, with or without associated anterior-chamber anomalies. Persistent hyaloid vessels, lens dislocation, iris dysplasia, aniridia, coloboma, microphthalmia, ocular calcifications, and optic disc pallor have also been reported. The most characteristic scalp lesion in ECCL is nevus psiloliparus: a soft, subcutaneous mass with a demarcated area of alopecia. Dermal lesions of the face included multiple papular or polypoid nodules. These lesions are unilateral in most cases; however, they can be bilateral occasionally. CNS lesions include cerebral and spinal lipomas, intracranial calcifications, abnormal intracranial vessels, ventricular and subarachnoid enlargement, agenesis of the corpus callosum, intracranial cysts, cortical dysplasia, and brain atrophy3). Jaffe-Campanacci syndrome (JCS) is also a rare syndrome characterized by multiple nonossifying fibromas of the long bones and jaw associated with café-au-lait spots and axillary freckles without accompanying neurofibromas4,5). In both of these rare diseases, neither short stature nor growth hormone (GH) deficiency has been previously reported. Here, we introduce the case of a 9-year-old Tajikistani girl with typical features of both ECCL and JCS in addition to GH deficiency.
Case report
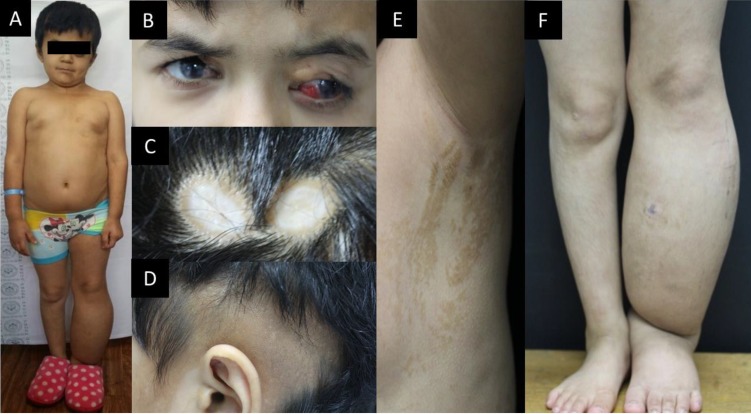
A 9-year-10-month-old Tajikistani girl visited our hospital through a missionary program. Her height was 110 cm, which was under the 3rd percentile for her age, and her weight was 23.1 kg, which was in the 5th–10th percentile. Her birth weight was 3.0 kg, which was in the 50th–75th percentile. She had incomplete upper eyelids (coloboma with ectopic cilia on the left side and cryptophthalmos with dermolipoma on the right) and sparse hair with separated alopecia. She had hyperpigmentation of the left hemibody with multiple café-au-lait macules on the left side of her face, trunk, and left extremity, and she had ipsilateral edema of the lower extremity. Her Tanner stage was Sexual Maturity Rating (SMR) 1. Her teacher mentioned that she displayed 2 years of retarded development compared with other children of the same age. She had no history of seizures. A grade I–II systolic heart murmur was heard on auscultation of the chest. Her family history was nonspecific. Her morphologic characteristics are shown in Fig. 1.
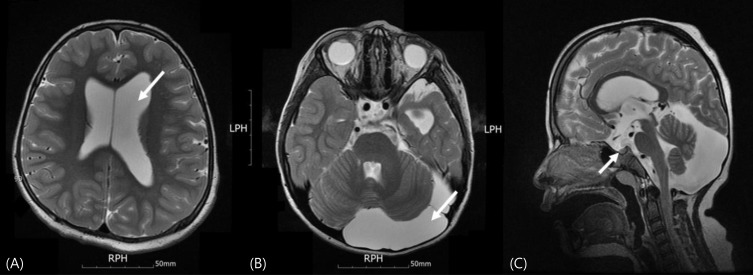
On the magnetic resonance imaging (MRI) of her brain, unilateral ventricular dilatation and an extra-axial cerebrospinal fluid space in the left hemisphere and posterior fossa were observed (Fig. 2), consistent with a minor MRI criterion for diagnosis of ECCL. Coarctation of the aorta (isthmic portion) with a velocity of 2.3–2.4 m/sec, which did not require any intervention, was observed on the 2-dimensional echocardiogram. Three major systems were involved and two major and two minor criteria, as defined by Moog6), were met. Thus, she was diagnosed with definitive ECCL.
The baseline Insulin-like growth factor-binding protein 3 (IGFBP-3) (1,245 ng/mL) and Insulin-like growth factor 1 (IGF-1) (19.14 ng/mL) were under the normal range (IGFBP3 normal range, 1,389–4,260 ng/mL; IGF-1 normal range, 76–499 ng/mL). The levels of the other measured hormones (T3, T4, free T4, thyroid-stimulating hormone, follicle-stimulating hormone, luteinizing hormone, adrenocorticotropic hormone, cortisol, estradiol, progesterone, testosterone, aldosterone, and renin) were within the normal range.
Her bone age was estimated to be 8 years 4 months; this was significantly less than her chronological age of 9 years 10 months. To evaluate her short stature, we performed a GH provocation test using insulin and levo-dihydroxyphenylalanine and found GH deficiency (Table 1). MRI of the sella was not performed; however, the abnormal findings on the brain MRI were not associated with GH deficiency (Fig. 2).
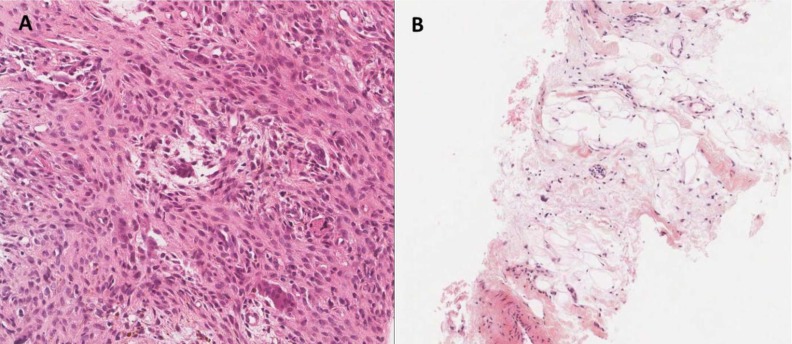
To evaluate the lesions to her extremities, we performed X-ray and MRI (Fig. 3). Images of her extremities showed nonossifying fibromas involving the left proximal humerus, left distal femur, both proximal tibias, and left proximal fibula, suggesting JCS4). Biopsy of the left tibia proximal bone and pretibial soft tissue were performed (Fig. 4). Pathologically, the results suggested nonossifying fibromatosis. Our present patient could be diagnosed with JCS because she had multiple café-au-lait macules and nonossifying fibromas, and no neurofibromatosis type-1 (NF1) mutation was found after genetic examination using a blood sample. Ultimately, she was diagnosed with GH deficiency combined with ECCL and JCS.
Discussion
ECCL is a rare, congenital neurocutaneous syndrome resulting from ectomesodermal dysgenesis and characterized by unique hairless scalp lesions in the form of nevus psiloliparus, ipsilateral ocular malformations, and CNS anomalies7). The genetic mechanism underlying these complex birth defects has been hypothesized to result from the action of lethal autosomal dominant genes and surviving because of mosaicism. Although the exact embryologic defect responsible for ECCL is unknown, the embryonic origin of the segmental choristomas and hamartomas is thought to be neural crest-derived mesectoderm. The causal role of anomalous neural crest morphogenesis and migration in the development of these abnormalities is supported by the report of a rare case of neurocutaneous melanosis in association with ECCL8). Several other hypotheses, such as maternal viral infection during pregnancy, and dysgenesis of the cephalic neural crest and anterior neural tube, have been proposed by previous authors9).
JCS is an ill-defined, rare syndrome associated with multiple nonossifying fibromas, skin manifestations (café-au-lait spots and axillary freckles) that has unclear interactions with NF1, and a genetic profile has not yet been determined4). She was prepubertal state. Her sex hormones were in the normal range. But her physical examination and sex hormones should be monitored about hypogonadism through puberty because hypogonadism could appear at JCS10).
Neurofibromas were not found on her body and her karyotype was normal. NF1 was ruled out because her clinical features were not consistent with this diagnosis, except for the presence of café-au-lait spots and intellectual disability. The majority of patients with café-au-lait macules and nonossifying fibromas or giant cell lesions harbor a pathogenic germline NF1 mutation, suggesting that many patients with JCS may actually have NF1. Stewart et al.11) found that 13 of 14 individuals with “classical” (multiple café-au-lait macules and multiple nonossifying fibromas or giant cell lesions) JCS harbored pathogenic mutations in NF1. Patients without classical JCS but with café-au-lait macules and a single nonossifying fibroma of a long bone or a giant cell lesion of the jaw also met the diagnostic criteria for NF1. Furthermore, all the examined patients had a pathogenic mutation in NF1.
Our patient was a rare example of combined ECCL and JCS. Her height was under the 3rd percentile and a GH deficiency was found. The growth of patients was not mentioned by Moog et al.6). Most reports of ECCL and JCS have described only morphologic lesions and were published in ophthalmology, dermatology, and orthopedics journals3,12). There are no reports about growth or development in patients with ECCL or JCS, with no discussion of whether GH therapy might help their growth. If a patient with ECCL or JCS has short stature, a study of the GH status and growth curve should be performed through long-term monitoring.