An 11-month-old girl with central precocious puberty caused by hypothalamic hamartoma
Article information

Abstract
Central precocious puberty (CPP) is caused by premature activation of the hypothalamic-gonadal axis, and must be treated adequately. In particular, CPP that occurs at a relatively young age or in boys is likely to be caused by an organic lesion. Hypothalamic hamartoma (HH) is the most common organic cause of CPP. The present case report describes an 11-month-old female infant who presented with vaginal bleeding and rapidly progressive secondary sex characteristics from the age of 6 months. She was diagnosed with CPP following the detection of HH via magnetic resonance imaging. The infant girl was successfully treated with gonadotropin-releasing hormone agonist. After 6 months, her breast had regressed and clinical and radiological follow-up demonstrated stable findings with no evidence of tumor growth or secondary sexual characteristics until the fourth year after the initiation of treatment. This patient is the one of the youngest infants presenting with CPP and HH in Korea; treatment was successful over a relatively long follow-up period.
Introduction
Precocious puberty (PP) is defined as the onset of the clinical signs of puberty at an earlier age, including breast budding before the age of 8 years in girls and testicular enlargement before 9 years in boys. Central precocious puberty (CPP) results from premature activation of the hypothalamic-pituitary-gonadal axis.
Of the cases of idiopathic CPP, >90% occur in girls and <10% in boys1). Common organic causes of CPP are hypothalamic hamartoma (HH), hydrocephalus, tumors, infections, congenital defects, ischemia, radiation, and brain injury.
HH, the most common cause of CPP, involves congenital malformation of the central nervous system. Patients with CPP exhibit symptoms such as epileptic syndrome (including gelastic seizure), PP, progressive cognitive decline, and behavioral disorders. In particular, 75% of PP cases that occur at a younger age (1–3 years) are due to HH; therefore, younger patients with PP require evaluations of the central causes of PP2).
In this case report, we describe an 11-month-old female infant with CPP caused by HH who presented with vaginal bleeding and rapidly progressive secondary sexual characteristics. She was treated with gonadotropin-releasing hormone (GnRH) agonist, which resulted in successful suppression of pubertal progression.
Case report
An 8-month-old infant girl presented with bloody vaginal discharge. She was born vaginally after 37+4 weeks of gestation with a birth weight of 2.94 kg and was the first child of healthy parents. Her mother's and father's height measurements are 160 and 168 cm, respectively. Her mother's menarcheal age was 13 years and her father experienced normal pubertal development. She was healthy until 2 months before admission. At around 6 months of age, bloody discharge was noted in her diaper, and 1 week previously, mucoid bloody discharge was present for 5 days. Her parents visited a local clinic and pelvic ultrasonography was performed, which showed an endometrial thickness of 17 mm and a cyst in the uterine appendage. She was referred to our hospital for further evaluation.
At the time of her first visit, her height was 67.0 cm (–1.42 standard deviation score [SDS]), weight 7.9 kg (–0.66 SDS), bone age 2–2+6 years, breast Tanner stage II, and pubic hair Tanner stage II. On her physical examination, no other abnormalities were observed. Laboratory tests demonstrated an elevated serum estradiol level of 37.4 pg/mL; basal luteinizing hormone (LH) and follicle stimulating hormone (FSH) levels were 4.0 and 4.4 IU/L, respectively. No considerable findings were detected during pelvic ultrasonography. The patient was scheduled for a follow-up after 3 months to examine pubertal progression.
At 11 months of age, the patient experienced 2 episodes of bloody vaginal discharge during the previous 3 months and her breasts progressed to Tanner stage III. At that time, her height was 77.0 cm (0.75 SDS) and she weighed 9.1 kg (–0.24 SDS). Her basal serum LH, FSH, and estradiol levels were 1.3 IU/L, 0.9 IU/L and 80.8 pg/mL, respectively. Thyroid function tests showed no specific abnormalities. On the GnRH stimulation test, peak LH and FSH levels were 65.9 and 12.1 IU/L, respectively, which was compatible with a diagnosis of CPP. The other results from the initial hormonal evaluation were within the normal limits for her chronological age. Brain magnetic resonance imaging (MRI) was performed to elucidate the organic cause, which demonstrated a well-defined isodense 1-cm-sized nonenhanced mass in the tuber cinereum on T1- weighted imaging, and this finding corresponded with HH (Fig. 1). Abdominopelvic computed tomography showed an enlarged ovary size for her chronological age (Fig. 2). As a result, the patient was diagnosed with CPP caused by HH. Her electroencephalogram demonstrated no epileptiform discharge.
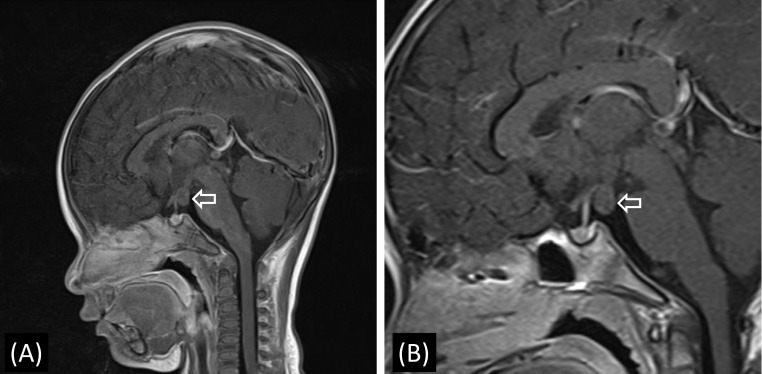
Magnetic resonance imaging findings of the patient. (A) T1-weighted sagittal image obtained at diagnosis showing a hypothalamic hamartoma (diameter: 1.0 cm) at the tuber cinereum with no enhancement (arrow). (B) T1-weighted sagittal image obtained at the age of 4+11 years showing no interval changes (arrow).
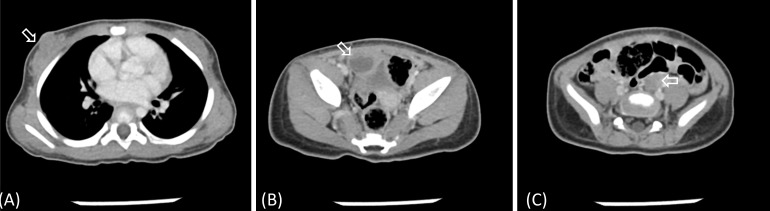
Abdominopelvic computed tomography findings of the patient obtained at diagnosis. (A) Breast parenchyma was predominantly observed (arrow). (B, C) Both ovaries were observed to be predominantly large for her age (B: 1.9 cm × 1.6 cm [arrow]; C: 1.7 cm × 1.0 cm [arrow]).
Following diagnosis, a GnRH agonist (Leuplin® depot, Takeda Pharmaceutical Co. Ltd., Osaka, Japan) was administered subcutaneously every 4 weeks. After 6 months, her breasts had regressed to Tanner stage I and her serum plasma estradiol level was <5 pg/mL. Subsequent tests for GnRH stimulation, peak LH, FSH and estradiol levels demonstrated that the levels had fallen to 5.0 IU/L, 5.0 IU/L, and <5.0 pg/mL, respectively. Following 1 year of GnRH agonist treatment, the patient's breasts remained at Tanner stage I and peak LH, FSH, and estradiol levels further decreased to 3.8 IU/L, 2.3 IU/L, and <5.0 pg/mL, respectively (Tables 1, 2).
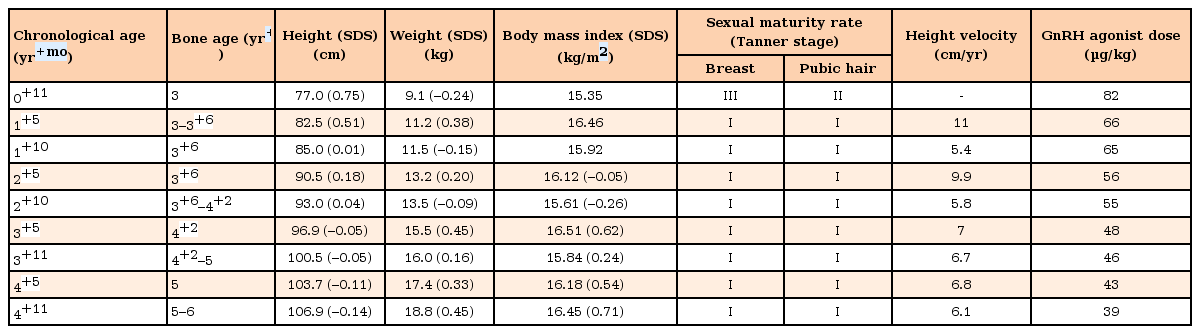
Clinical course of the patients
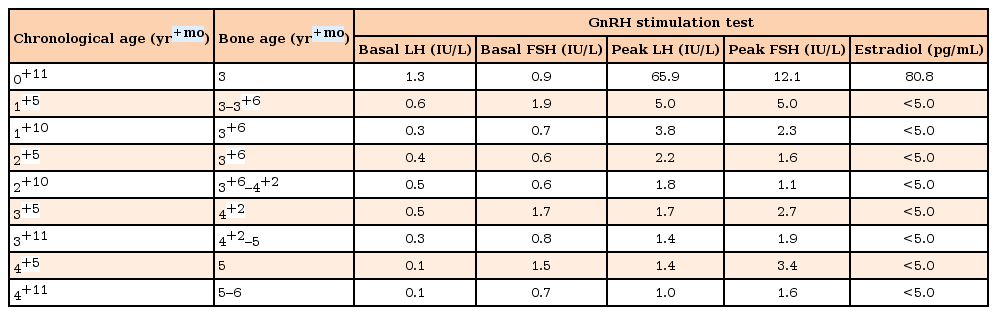
Gonadotropin-releasing hormone (GnRH) stimulation test during follow-up
The duration of follow-up was 4 years. Following the initiation of GnRH agonist therapy, no further secondary sexual characteristics or vaginal bleeding were observed. Serum peak LH levels after GnRH stimulation testing were well suppressed. Height velocity was maintained at 6–7 cm per year and her bone age is consistent with her chronological age to date (Tables 1, 2). Adverse events of GnRH agonist treatment were not noted. Symptoms and signs associated with HH, such as seizures, psychological problems, and cognitive decline were not observed. Follow-up brain MRI imaging at age 1 and 4, demonstrated that the HH did not exhibit any changes in size or contouring during GnRH agonist treatment (Fig. 1).
Discussion
HHs are predominantly located at the base of the hypothalamus where they develop as an outgrowth of the floor of the third ventricle. HH are rare congenital non-neoplastic lesions containing mature tissue3). The incidence of HH is not well documented; however, it is the most common organic cause of CPP in both boys and girls. Several studies have reported the incidence of HHs in patients with isosexual PP, with values ranging from 14% to 58%4).
HH has a triad of symptoms, including gelastic seizure, PP, and behavior and cognitive disorders. The existence of symptoms or their severity is variable between patients. Patients with PP typically present with an onset of pubertal development at a very young age usually <4 years of age5). Therefore, relatively early presentation of PP or boys with PP requires clinical evaluation for brain tumors, such as HH.
Han et al.6) analyzed the clinical characteristics and outcome of 11 patients with HH in a single center in Korea for 11 years from 1986. The mean age at symptom presentation was 5 years and 8 month (range, 1.1–14.1 years), with symptoms including gelastic seizure (n=7, 64%), PP (n=6, 55%) and behavioral or psychological disorders (n=9, 82%). Seizure symptoms were not responsive to medical treatment and were successfully controlled by surgery. Lee et al.7) also analyzed 11 HH patients in Korea's single medical center for 13 years, and PP was detected in 7 patients (64%) and seizure in 7 patients (64%). Among the 7 patients with PP, PP was demonstrated to be the first symptom of HH. The mean age of symptom onset was 5.4 years. Debeneix et al.8) analyzed 19 patients with HH for 17 years in France. The mean age at diagnosis was 5.7±4.1 (standard deviation) years. CPP was detected in 14 patients (73.6%), seizure in 9 patients (47.4%), isolated CPP in 9 patients, and CPP and seizure in 4 patients, respectively. Notably, 16 of 19 patients exhibited no changes in the contour and size of the HH after a mean follow-up period of 4.2±3.3 (0.5–10.2) years.
The optimal tool for diagnosis of HH is high-resolution brain MRI. Symptoms of HH are closely correlated with the position of HH and its connections to surrounding tissue, such as the hypothalamus. Seizures commonly occur in patients with HH that infiltrate the hypothalamus as opposed to in patients with pedunculated HH9). However, >45% of HH are mixed types that exists in the third ventricle and extends inferiorly into the interpeduncular fossa, and in this case, both seizures and PP were observed in the patients10). Patients with pedunculated HHs without infiltrating hypothalamus, which are those located below the third ventricle, rarely present with seizures. Rather, these lesions are commonly associated with CPP11). The present patient was thought to possess a pedunculated HH as she exhibited isolated PP. However, regular follow-up is warranted for this patient, since seizure or behavior associated with a cognitive disorder might occur.
The apparent association between HH and CPP is still not completely understood. Judge et al.12) reported that luteinizing hormone-releasing hormone (LHRH) immunoreactivity was detected in surgically removed HH, indicating that the HH has a heterotopic LHRH pulse generator and follow puberty. Boyko et al.13) and Striano et al.14) reported that a pedunculated HH compresses the pituitary stalk, interrupting the inhibitory pathways from the hypothalamus between the pituitary posterior lobe. Consequently, excessive secretion of GnRH is transferred to the hypothalamus through the tuber cinereum, which provokes abnormal endocrine function.
In infancy, elevation of LH and FSH levels is referred to as mini puberty; symptoms such as vaginal bleeding or breast budding might be observed transiently15). Previously reported cases of infants with vaginal bleeding were born preterm or were small for their gestational age. These extreme cases of mini puberty detailed only 1–2 episodes of vaginal bleeding1617). In girls, postnatal gonadotropin levels started to decrease after the peak at 1–3 months of age. However, the present patient was born full-term and was of the appropriate size for gestational age; she experienced vaginal bleeding for 5 days and more than 3 times. Moreover, our patient showed rapid progression of her secondary sexual characteristics during the follow-up period and brain MRI revealed HH. Therefore, we concluded that her pubertal progression was not caused by mini puberty but by HH.
Although the optimum treatment of HH is surgical removal, complete removal remains difficult owing to the adhesion or invasion of surrounding tissues. Patients with uncontrolled seizures sometimes require surgery. CPP due to HH is well controlled by hormonal therapy. GnRH agonist treatment induces persistent sensitization of the GnRH receptor, followed by decreased sensitivity and the expression of the receptor as a result of delayed puberty18). Lee et al.7) reported that 6 PP patients with HH were diagnosed before 7 years of age and treated at the appropriate time. Only one patient demonstrated pubertal sign at 10 years 6 months; however, treatment with the GnRH agonist was initiated due to prominent LH concentration on GnRH stimulation test and the concern about attenuated adult height and emotional distress. Kotwal et al.19) reported the case of a 16-month-old girl who had shown vaginal bleeding from when she was 6 months old, which evolved into a monthly regular cycle and progressive secondary sexual characteristics. The researchers detected HH in her brain MRI. She was treated by GnRH analogue and has shown a good response on two-year follow-up. Acharya et al.20) reported a 4-month-old male infant with CPP due to HH diagnosed in utero. He had a history of accelerated growth since birth and developed secondary sexual characteristics from 3 months of age. To exclude mini puberty, the patient was followed-up after 4 months; his bone age was 18 months and GnRH stimulation test exhibited PP. He was also treated with leuprolide acetate and his growth stabilized after 9 months of follow-up.
We report the clinical course and MRI findings during the 4-year follow-up period of the one of the youngest patients in Korea that exhibited PP as a first symptom of HH. This infant girl with CPP caused by HH exhibited a good response to the treatment with GnRH agonist. In conclusion, rapidly progressive CPP in infancy requires appropriate laboratory and radiologic evaluation to determine the organic cause, and should be distinguished from extreme mini puberty.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.