Hypotonic hyponatremia by primary polydipsia caused brain death in a 10-year-old boy
Article information
Abstract
Hypotonic hyponatremia by primary polydipsia can cause severe neurologic complications due to cerebral edema. A 10-year-and-4-month-old boy with a psychiatric history of intellectual disability and behavioral disorders who presented with chief complaints of seizure and mental change showed severe hypotonic hyponatremia with low urine osmolality (serum sodium, 101 mmol/L; serum osmolality, 215 mOsm/kg; urine osmolality, 108 mOsm/kg). The patient had been polydipsic for a few months prior, and this had been worse in the previous few days. A diagnosis of hypotonic hyponatremia caused by primary polydipsia was made. The patient was in a coma, and developed respiratory arrest and became brain death shortly after admission, despite the treatment. The initial brain magnetic resonance imaging showed severe brain swelling with tonsillar and uncal herniation, and the patient was declared as brain death. It has been reported that antidiuretic hormone suppression is inadequate in patients with chronic polydipsia, and that this inadequate suppression of antidiuretic hormone is aggravated in patients with acute psychosis. Therefore, hyponatremia by primary polydipsia, although it is rare, can cause serious and life-threatening neurologic complications.
Introduction
Hyponatremia is defined as a serum sodium level of less than 135 mmol/L, and is considered severe when the level is below 125 mmol/L. Hypotonic or dilutional hyponatremia, the most common form of hyponatremia, is caused when water intake exceeds the water excretion capacity1). Hypotonicity in hypotonic hyponatremia can cause cerebral edema, a potentially life-threatening complication of hyponatremia1). Fortunately, it has been demonstrated that the brain is relatively resistant to osmotic swelling, via defense mechanisms: first, fluid shift from the interstitial space of the brain into cerebrospinal fluid, and second, loss of cell solutes, primarily potassium, from the brain2). Through these defense mechanisms, if hyponatremia develops subacutely or chronically, acute neurologic symptoms of hyponatremia, which are mostly consequences of cerebral edema, do not occur2). Therefore, most patients with acute symptomatic hyponatremia are either hospitalized patients with postoperative hyponatremia, patients with water intoxication due to psychogenic polydipsia, or patients to whom thiazide diuretics have recently been prescribed, all of which can abruptly disrupt the patient's homeostasis2). Here we report a case of psychogenic polydipsia that caused severe hypotonic hyponatremia, which in turn resulted in severe brain swelling that eventually led the patient to brain death.
Case report
A 10-year-and-4-month-old boy (height 150 cm, 75-95 percentile, weight 67 kg, >97 percentile) was admitted to the intensive care unit via the emergency center with a chief complaint of mental change after one episode of generalized tonic-clonic seizure.
Prior medical history revealed that the patient had had one suspicious episode of seizure 6 years before, and had received an evaluation including blood tests, brain computed tomography (CT) scanning and electroencephalogram (EEG). All of the results were normal except for a subtle abnormality in the EEG, which was considered clinically insignificant. He had shown no more suspicious seizure events since that time without medication. He has been diagnosed with intellectual disability (intelligence quotient score of 44) and attention deficit hyperactivity disorder (ADHD) in a local psychiatric clinic. Medications included methylphenidate, atomoxetine, aripiprazole, fluvoxamine, benztropine, and topiramate on a daily basis. There was not any notable family history.
The physical examination revealed that the patient was in a comatose state, not responding to any noxious stimuli. Pupils were fixed and equal with a diameter of 5 mm each. Lung sounds were decreased on auscultation and the heart examination was normal. The abdomen was soft and not distended, and hypoactive bowel sounds were present. Rectal examination revealed hypotonic sphincter tone. Admission vital signs were as follows: pulse of 75 beats per minute, abnormally high blood pressure of 167/118 mmHg, respirations of 22 per minute, and temperature of 36.2℃.
Initial chest x-ray and chest CT scan showed findings compatible with pulmonary edema, and brain CT scan revealed severe brain swelling. Brain magnetic resonance imaging (MRI) showed severe brain swelling with tonsillar and uncal herniation (Fig. 1).
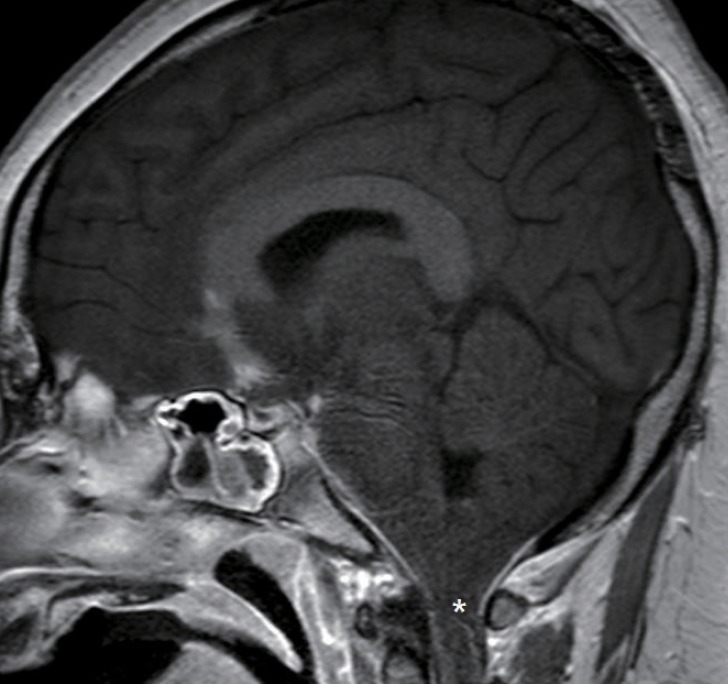
Brain magnetic resonance imaging showing severe brain swelling with tonsillar and uncal herniation (asterisk).
Although the laboratory findings 3 months prior to admission, performed when he visited our emergency center due to cervical lymphadenopathy, showed no abnormal findings, on the admission day, severe hypo-osmolar hyponatremia (serum Na, 101 mmol/L [basal reference values {BRV}, 135-145 mmol/L]; serum osmolality, 215 mOsm/kg [BRV, 289-308 mOsm/kg]) was found on the initial laboratory examination, along with low urine osmolality and low spot urinary sodium concentration (urine osmolality, 108 mOsm/kg [BRV, 50-1,200 mOsm/kg]; urine Na, 19 mmol/L). Serum glucose, total protein, albumin, blood urea nitrogen, and creatinine levels were all within normal range. The plasma adrenocorticotropin level was slightly low (5.54 pg/mL [BRV, 7.2-63.3 pg/mL]), while the serum cortisol level was in the normal range (12.5 µg/dL [BRV, 6.7-22.6 µg/dL]). Thyroid hormones were in the normal range (T3, 0.67 ng/mL [BRV, 0.58-1.59 ng/mL]; free T4, 0.97 ng/dL [BRV, 0.7-1.48 ng/dL]; thyroid-stimulating hormone, 2.97 µIU/mL [BRV, 0.35-4.94 µIU/mL]). The antidiuretic hormone (ADH) level was mildly elevated (13.34 pg/mL [BRV, 0-6.7 pg/mL]).
Shortly after admission, the patient experienced respiratory arrest. Endotracheal intubation was immediately done, and the patient was put on mechanical ventilation. After intubation, his blood pressure fell to 64/39 mmHg, and intravenous inotropics were administered. During the first 3 hours after admission, the patient showed polyuria with urination of 700 mL in 3 hours. After 3 hours, however, oliguria developed with sudden decrease in urination to 30 mL/hr even though diuretics were administered. The patient was put on continuous renal replacement therapy (CRRT) using continuous veno-venous hemodialysis.
After acute management, further past history was taken from the patient's mother, and it was revealed that for several months prior to admission, he had suffered from worsening of inattention, impulsivity, and hyperactivity, which led to him dropping out of school in the prior month. He also had difficulty controlling impulsive eating, and ate one type of food in excessive amounts for a period time. For example, he ate a lot of toothpaste for a while, and there was also a period of time when he ate a lot of soap. Since around 2 to 3 months prior to the event, his mother noticed that the patient drank excessive amounts of water, even drinking water directly from the shower tap, but she had not yet reported this change to the psychiatrist. The polydipsia had become worse in the previous few days, resulting in water regurgitation from his stomach when he lay down, as well as frequent bed-wetting.
Together with the patient's history and laboratory results, we concluded that the patient was in hypotonic hyponatremia due to psychogenic polydipsia.
For the treatment of brain edema, intravenous mannitol was administered. For hyponatremia, increment of serum sodium concentration was done by administration of hypertonic saline solution and intravenous furosemide, and fluid restriction. Using 3% sodium chloride fluid and normal saline solution together, 5.5 mEq/kg of sodium was first administered, and the sodium concentration of the fluid and fluid balance target were continuously adjusted according to the patient's serum electrolytes that were measured every 2 to 4 hours. The patient's serum sodium level had increased from 101 to 108 mmol/L by 8 hours after admission, and the subsequent serum sodium concentration were 110 mmol/L at 24 hours after admission, 117 mmol/L at 48 hours after admission, 127 mmol/L at 72 hours after admission, and reached the normal range at 96 hours after admission at 139 mmol/L (Table 1).

Serial laboratory data
On the third day of his admission, a sudden increase in urine output (>350 mL/hr) occurred, with an increase in serum osmolality (291 mOsm/kg) and a decrease in urine osmolality (67 mOsm/kg). With the impression of central diabetes insipidus (DI), desmopressin acetate (Minirin, Ferring pharmaceuticals, Saint-Prex, Switzerland) was subsequently started, and CRRT was discontinued. The patient remained on desmopressin acetate after that, and his electrolytes were kept stable.
An EEG was performed on the fourth day of admission, and showed extremely low voltage and featureless background rhythms. The patient did not show self-respiration during an apnea test. On the 20th day of admission, after thorough neurological examinations together with the results of the EEG and brain MRI, the patient was declared to be in a state of brain death by a panel of neurologists.
Discussion
Hypotonic hyponatremia most commonly occurs in patients with impairment of renal water excretion, but, in a minority of cases, it is caused by excessive water intake, with normal or nearly normal renal excretory capacity1). Such cases include primary polydipsia, use of diluted infant formula, use of sodium-free irrigants or solutions, and multiple tap-water enemas1).
Primary polydipsia, also called psychogenic polydipsia, hysterical polydipsia, compulsive polydipsia, dipsomania, and potomania, appears most commonly in chronically mentally ill populations, and an epidemiological surveys has shown that more than 20% of chronic psychiatric inpatients were polydipsic3). Primary polydipsia most commonly occurs in schizophrenic patients, but other disorders including pseudomania, delusional depression, bipolar disorder, chronic alcoholism, self-induced vomiting, mental retardation, autism, severe personality/behavioral disorder, and dementia are also associated4). In our case, the patient was diagnosed of mental retardation and ADHD and these disorders can be the causes of polydipsia. However, the reason for recent worsening of his behavioral symptoms could not be investigated further, as the patient presented in comatose state and progressed to brain death.
The underlying causes of primary polydipsia are still under investigation, and it is speculated that multiple mechanisms contribute to cause polydipsia. One theory is that polydipsic patients have increased drive for fluids in a sense of addiction, as the affinity of opioid agonists to their receptors are increased up to 50 times in hyponatremic environment56). Other theories include hyperactivity of hypothalamic thirst regulation centers, and hippocampal dysregulation for repetitive behavior of consuming fluids78).
Antipsychotics have also been speculated as the cause for the onset or worsening of polydipsia. However, antipsychotics solely cannot explain polydipsia, as there have been cases of patients with polydipsia who had not been taking medication9). In our case, psychiatrists reviewed the patient's history under consultation, and gave the opinion that the probability of his medications being the cause of polydipsia was low, as he had been taking the medications without any change for a long time and did not complain dry mouth.
Polydipsia-hyponatremia syndrome, the condition in which polydipsia results in hyponatremia, occurs in 5% to 10% of patients with primary polydipsia4). It can be caused by combination of several factors. One contributing factor is the severity of polydipsia, which result in consumption of fluids overwhelming the capacity of nephrourinary system3). Presence of syndrome of inappropriate secretion of ADH (SIADH) or osmotic dysregulation in polydipsic patients has also been reported as a contributing factor10). In these patients, free water clearance was lower and urine osmolality values were higher than in controls after water loading10). In addition, plasma ADH levels in patients with polydipsia were higher than those in controls for concurrent levels of plasma osmolality and serum sodium, and the osmotic threshold for ADH secretion was also reduced in patients with polydipsia410). In other words, ADH in patients with polydipsia began to be suppressed at lower levels of plasma osmolality and serum sodium than in controls. One hypothesis for the presence of SIADH in polydipsic patients is that chronically increased intravascular volume in these patients resulted in the resetting of the osmostat. This reduced osmotic threshold for ADH, in turn can lead to inadequate ADH suppression causing decrease in free water excretion4). Besides, Goldman et al.10), suggested that the renal sensitivity to ADH is increased in polydipsic patients. Either way, polydipsia combined with inadequately high ADH levels or efficacies can cause severe hypotonic hyponatremia in polydipsic patients. In our case, the patient drank excessive amount of fluids that could have overcome the renal excretory capacity. In addition, the initial plasma ADH level of the patient was 13.34 pg/mL, which was relatively high considering his condition of severe hypo-osmolar hyponatremia. The patient in our case had been polydipsic for several months, and it is possible that his ADH secretion had been affected. Central DI was also present in our case during the clinical course on the 3rd day of admission. The reason for this is brain death due to hypotonicity that led to brain herniation. Brain death is one of the most common causes of DI in children due to absence of cerebral circulation causing inability of ADH production11). The carotid Doppler sonography of our patient showed absence of significant blood flow in bilateral internal carotid arteries.
Emsley et al.12) also conducted water-loading test in patients with schizophrenia or schizoaffective disorder and in healthy controls. In addition to diminished urine output and higher minimum urine osmolalities in psychotic patients, they also noted that patients who had been acutely psychotic for less than 6 months showed the most severe antidiuretic state and had the highest plasma ADH levels12). Further impaired urinary dilution and urine excretion in acute psychosis, therefore, is probably the reason for water intoxication in primary polydipsia. In our case, too, the patient had shown worsening of behavioral disorders including inattention, impulsivity, and hyperactivity. This could also have attributed to aggravation of hyponatremia.
Despite its potential threat, polydipsia in chronically psychotic patients is often undetected until neurologic symptoms develop, as in our case, because such histories are difficult to obtain from patients. Therefore, careful observation by caregiver is important. Clues for development of polydipsia are preoccupation with drinking, nocturnal bed wetting, urinary incontinence, and worsening of psychosis in the afternoon and early evening4). Once the polydipsia is detected, interventions such as fluid restriction according to diurnal and morning to afternoon weight changes, behavioral interventions using biofeedback approach, and pharmacological interventions can be preceded to modulate polydipsia and prevent symptomatic hyponatremia413).
In conclusion, though it is rare, psychogenic polydipsia can cause severe hyponatremia leading to fatal neurologic consequences. In this case, the consequences were severe cerebral edema and brain herniation causing seizure, DI, and eventually, brain death.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.