Introduction
Neurohypophyseal (central) diabetes insipidus (DI) is characterized by deficient or absent secretion of arginine vasopressin (AVP), resulting in polyuria and polydipsia1). It is typically caused by tumors, infection, or trauma2).
Familial neurohypophyseal DI is a rare inherited disorder accounting for 1%-5% of all cases of central DI. The majority of cases are inherited in an autosomal dominant pattern3,4). Familial neurohypophyseal DI is caused by a mutation in the 2.5 kb AVP-neurophysin II (AVP-NPII) gene located on chromosome 20p135). Since the first AVP-NPII mutation was reported6), 62 additional mutations have been identified1,7).
In this study, we report a case of familial neurohypophyseal DI caused by a mutation (c.286G>T) in the AVP-NPII gene in four generations of a Korean family.
Case report
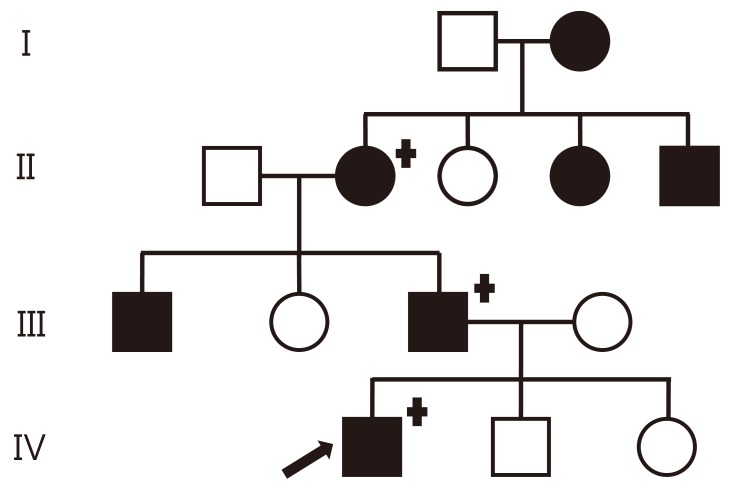
A 7-year-old boy was admitted to the hospital for persistent symptoms of polydipsia and polyuria that started in early infancy. He drank an average of 3.9 L of water per day and urinated an average of 5.1 L per day, including at least three times during the night; on physical examination, he was otherwise healthy. His height and weight were 123.3 cm (39th percentile) and 20.7 kg (15th percentile), respectively. His medical history was unremarkable, and he did not have a history of head trauma or symptoms of neurological or pituitary dysfunction. An evaluation of the family history revealed seven family members, spanning four generations, who were suspected of having the same symptoms for an extended period of time (Fig. 1). His father reported an intake of 11 L of water per day and a urine output of 12 L per day. His grandmother reported an intake of 10 L of water per day and a urine output of 9 L per day. The pedigree was consistent with autosomal dominant inheritance.
The basal plasma levels of free thyroxine, total triiodothyronine, thyroid stimulating hormone, cortisol, adrenocorticotropic hormone, insulin like growth factor-I (IGF-I), IGF binding protein-3 were within the normal ranges. The basal plasma osmolarity was 278 mOsm/kg H2O, the urine osmolarity was 55 mOsm/kg H2O, the urine specific gravity was less than 1.005, the plasma sodium was 139 mEq/L, and the antidiuresis hormone (ADH) level was 8.66 pg/mL. He underwent a standard water deprivation test of sufficient duration to increase plasma osmolarity and plasma sodium above 300 mOsm/kg H2O and 145 mEq/L, respectively. After seven hours, the plasma osmolarity and sodium increased to 302 mOsm/kg H2O and 145 mEq/L, respectively; the urine osmolarity was 349 mOsm/kg H2O, and the ADH was 5.62 pg/mL. During the test, he continued to have inappropriately dilute urine and fluid diuresis. His body weight decreased by 9%. After a desmopressin challenge test of 20 ┬Ąg, his urine osmolarity increased to 621 mOsm/kg H2O. These results confirmed a diagnosis of partial vasopressin-deficient DI (Tables 1,2).
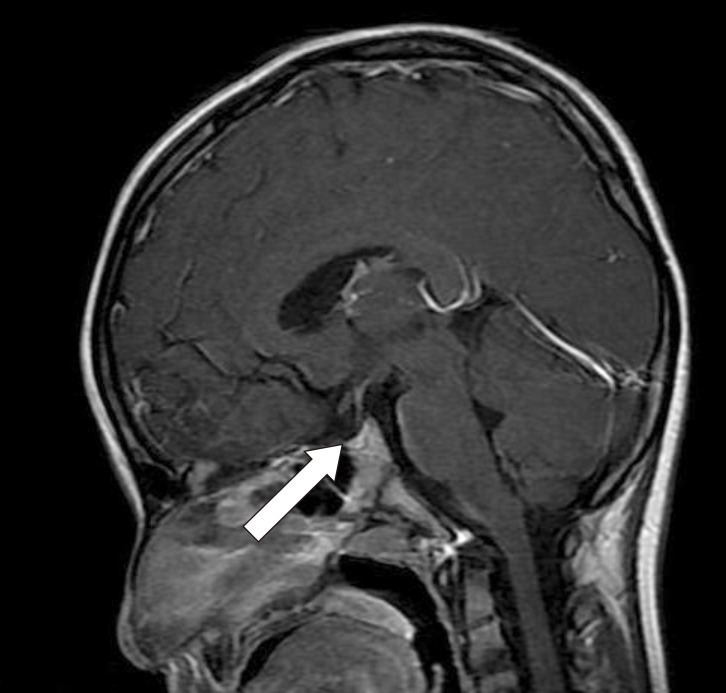
The pituitary magnetic resonance imaging (MRI) showed a relatively small and hypointense neurohypophysis (Fig. 2).
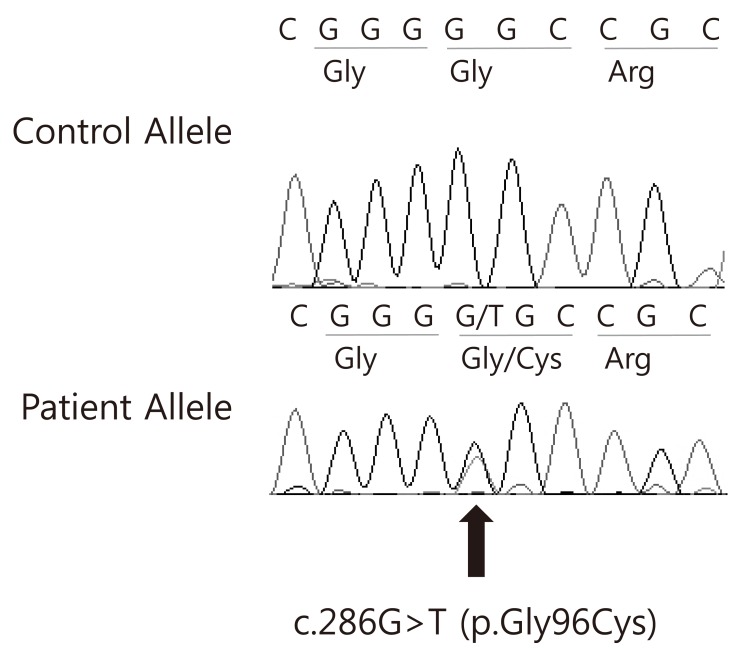
After informed consent was obtained, peripheral blood specimens were collected from the his family members for molecular genetic analysis. Genomic DNA (gDNA) was extracted using the Wizard genomic DNA purification kit according to the manufacturer's instructions (Promega, Madison, WI, USA). All coding exons and their exon-intron boundaries of the AVP gene were amplified by polymerase chain reaction using primers designed by the authors (available by request). Cycle sequencing was performed with a Big Dye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3130xl Genetic Analyzer (Applied Biosystems). The sequencing results were compared to a reference sequence of the AVP gene, GenBank accession number NM 000490.4. The sequence variation was described according to the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/). Direct sequencing analysis of the AVP-NPII gene revealed a heterozygous c. 286G>T mutation predicting an amino acid substitution (Gly96Cys) in exon 2. The AVP-NPII genes of the his father and grandmother were found to have the same mutation (Fig. 3).
After treatment with oral desmopressin 0.1 mg two times daily was started, the symptoms of polydipsia and polyuria were satisfactorily controlled, resulting in a dry bed throughout the night, and an improvement in food intake and growth. In three months, he gained 3 kg. Currently he drinks approximately 1.5 L of water daily and has a urine output of 1.5 L per day. His father and grandmother's water intake/diuresis per day are 1.8 L/1.8 L and 3.0 L/2.0 L, respectively, due to the effects of the medication.
Discussion
Neurohypophyseal DI is a rare disorder of water conservation resulting from a deficiency of AVP. Under normal circumstances, the precursor hormone prepro-AVP is produced by neurosecretory cells in the supraoptic and paraventricular nuclei of the human hypothalamus8). During axonal transport to the pituitary gland, prepro-AVP is cleaved to become AVP, and it is finally stored in the posterior pituitary gland (neurohypophysis). When water deprivation causes a rise in plasma osmolarity above 280-290 mOsmol/kg H2O, AVP is released into the circulation. In the collecting ducts of the kidney, AVP binds to the vasopressin type 2 receptor, inducing upregulation of the aquaporin II channels. This process results in increased water retention with a rise in urine osmolarity to a maximum of 1,000-1,200 mOsmol/kg H2O and a restoration of plasma osmolarity to within the reference range1,9).
Familial neurohypophyseal DI is rare disorder accounting for only 1%-5% of all causes of central DI2,10). Familial neurohypophyseal DI typically presents between the ages of 1 and 6 years with polyuria and polydipsia and is radiologically characterized by the loss of the posterior pituitary bright spot on T1 MRI, suggesting dysfunction of the neurohypophysis11).
Familial neurohypophyseal DI is caused by mutations in the AVP-NPII gene, which are usually inherited in an autosomal dominant pattern12). The AVP-NPII gene is located on chromosome 20p13 and has three exons, with an open reading frame of 492 bp. Exon 1 encodes a signal peptide, AVP, and the N-terminal portion of NP II; exon 2 encodes the central region of NP II; and exon 3 encodes the C-terminal part of NP II and copeptin, a glycoprotein with an unknown function8,13). The mutations involved in familial neurophyphyseal DI include small deletions, as well as missense and nonsense mutations that affect the signal peptide, the AVP moiety, or the AVP carrier protein, NPII. The majority of these mutations have been found in the region of the gene encoding NPII, an intracellular binding protein for AVP. Only a few mutations have been localized to the signal peptide or the AVP coding sequence, and no mutations have been localized to the glycoprotein moiety. It is assumed that all known autosomal dominant mutations cause defective folding or dimerization of the precursor protein14,15).
Since the first AVP-NPII mutation was reported6), 62 different mutations have been identified1,7). In Korea, a splice site mutation within the intron, a missense mutation in exon 2 (+1692C>A), and a deletion mutation of Glu78have been reported5,10).
In this study, we identified a Korean family with autosomal dominant familial neurohypophyseal DI in four generations, and we identified the first heterozygous missense mutation in exon 2 of the AVP-NPII gene (c.286G>T) in Asia. The proband's father, uncle, grandmother, grandmother's sister, grandmother's brother, and great-grandmother each have a long history of polyuria and polydipsia, suggesting autosomal dominant transmission of the disease. Additionally, the proband's great-grandmother might have been the first person affected and thus would be expected to have milder signs, although she was not tested for the disease. The diagnosis was confirmed in the proband by a water deprivation test and in three family members (proband, proband's father, proband's grandmother) by DNA analysis. Genetic analysis of the AVP-NPII region revealed a heterozygous missense mutation in exon 2 of the AVP-NPII gene (286G>T), and this amino acid substitution (Gly96Cys) was predicted to have occurred in the three clinically affected subjects. The transition G286T, causing substitution of Gly96Cys, was previously reported in a case of familial neurohypophyseal DI in the United States16).
In summary, we report a Korean family with autosomal dominant familial neurohypophyseal DI affecting four generations with a heterozygous missense mutation in exon 2 of AVP-NPII (c.286G>T, p.Gly96Cys). The presence of this mutation suggests that the portion of the neurophysin peptide encoded by this sequence is important for the appropriate expression of vasopressin.