Introduction
Cushing syndrome is defined as the clinical manifestations resulting from prolonged exposure to excessive glucocorticoid, whereas Cushing disease specifies Cushing syndrome that is caused by excessive secretion of adrenocorticotropic hormone (ACTH) from a pituitary tumor, usually an adenoma1).
The incidence of Cushing disease is very rare in childhood and adolescence, and endocrine dysfunctions such as growth failure and delayed or precocious puberty are more common than in adults2). Cushing disease accounts for approximately 75%-80% of all cases of endogenous Cushing syndrome in children over 7 years of age2,3). Among 182 cases taken from the literature, the median age at presentation was 14.1 years, and the youngest reported case was 5.8 years4). In children younger than 7 years, adrenal causes of Cushing syndrome are the most common3).
Most children and adolescents with Cushing disease have a typical Cushingoid appearance. Also, height standard deviation score (SDS) is almost always reduced with increased body mass index (BMI). Bone age is typically delayed by about 2 years (range, -0.5 to 4.1 years)5). Because bone age delay is negatively correlated with height SDS, early diagnosis is essential to pediatric patients' future development. The clinical aspects of pediatric Cushing disease that differ from those in adults are as follows: high incidence in prepubertal males compared to females, low incidence of corticotroph adenoma on pituitary imaging, high frequency of lateralization of ACTH secretion demonstrated by bilateral inferior petrosal sinus sampling (BIPSS), and rapid response to external-beam pituitary radiotherapy6). The other clinical features of Cushing disease, namely hirsutism, abdominal striae, hypertension, emotional lability, fatigue, muscle weakness, and easy bruising, are less common in pediatric cases than in adults4).
In both adult and pediatric Cushing disease, the first-line treatment is selective microadenomectomy for preservation of normal pituitary tissue; this is accomplished by the transsphenoidal approach7). As for the long-term course of pediatric Cushing disease, there are, as reflects the rarity of such cases, few reports. Herein, we report the case of a 16-year-old boy with Cushing disease who presented with rapid weight gain and stunted growth velocity, and who was followed-up on for 10 years for management of a long-term clinical course after transsphenoidal surgery for pituitary microadenoma.
Case report
A 16-year-old male was referred to our institute Asan Medical Center Children's Hospital for evaluation of rapid weight gain and stunted growth velocity over 4 years. On admission, the patient's systolic and diastolic blood pressure was 108 and 72 mmHg, respectively. Body weight and height were 61.1 kg (-0.18 SDS) and 138.5 cm (-5.44 SDS), respectively. The BMI was 31.1 kg/m2 (+2.16 SDS). He displayed a moon-shaped face, buffalo hump, truncal obesity, acanthosis nigricans, and abdominal striae. The volume of each testis was 1 mL, while the Tanner stage of pubic hair was 2.
The serum cholesterol level was 173 mg/dL, and serum sodium and potassium levels were 144 mmol/L and 4.5 mmol/L, respectively. The serum insulin and glucose levels were 24.7 µU/mL and 74 mg/dL, respectively. Hemoglobin A1c was 5.8%. The serum cortisol levels in the morning and evening were 23.1 µg/dL and 26.2 µg/dL, respectively, suggesting loss of circadian rhythm. The 24-hour urine free cortisol level was elevated to 160 µg/day (<50 µg/day). The serum cortisol level after overnight dexamethasone suppression was 16.1 µg/dL (<5 µg/dL). A low-dose dexamethasone-suppression test was performed according to the standard protocol (oral dexamethasone 20 µg/kg/day for 2 days). The plasma ACTH, serum cortisol and 24-hour urine free cortisol levels were 40.6 pg/mL (<20 pg/mL), 29.4 µg/day (<5 µg/dL), and 73.5 µg/day (<10 µg/day), respectively, indicating a definitive diagnosis of Cushing syndrome. Subsequently, a high-dose dexamethasone test with an oral dexamethasone dose of 80 µg/kg/day demonstrated suppressed plasma ACTH (16.6 pg/mL) and serum cortisol (2.0 µg/dL) levels, indicating that the Cushing disease had been caused by pituitary ACTH secretion.
The patient's bone age, as determined by the Greulich-Pyle method8), was 11 years. A simple x-ray of the thoracolumbar spine showed osteopenia (Fig. 1), and bone mineral density (BMD) z-scores of the lumbar spine and femur neck were -6.127 and -4.518, respectively, as determined by dual-energy x-ray absorptiometry (Lunar Co., Madison, WI, USA). Sellar magnetic resonance imaging (MRI) did not show any evidence of pituitary tumor. As this result suggested a pituitary source of ACTH secretion, BIPSS was performed to determine the laterality of the ACTH secretion. The peak ACTH level measured from the right inferior petrosal sinus (IPS) after injection of corticotropin-releasing hormone (CRH) was remarkably higher than that measured from the left IPS, suggesting right lateralization of ACTH secretion (Table 1).
Subsequently, a right hemihypophysectomy was performed by the endonasal transsphenoidal approach. Examination of the biopsied tissue suggested pituitary microadenoma. However, the 24-hour urine free cortisol was persistently elevated to 85.8 µg/day. Repeated BIPSS, moreover, did not show lateralization of ACTH secretion. The peak ACTH levels, though, were markedly increased after CRH loading from both the left and the right petrosal sinus (Table 2). Fourteen days after the right hemihypophysectomy, a total hypophysectomy was performed by the transsphenoidal approach. The histological findings were suggestive of nonneoplastic pituitary tissue. Postoperatively, the 24-hour urine free cortisol was 22.5 µg/day, and the plasma ACTH and serum cortisol levels were 6.9 pg/mL and 1.2 µg/dL, respectively. Owing to symptoms of adrenal insufficiency, hydrocortisone were given at a stress dose of 100 mg/m2/day intravenously after surgery and subsequently tapered to the physiologic oral dose of 15 mg/m2/day.
An L-dopa test and combined anterior pituitary function test after 2 weeks of the total hypophysectomy revealed growth hormone (GH) deficiency, central hypothyroidism, hypogonadotropic hypogonadism, and ACTH deficiency (Table 3). Levothyroxine sodium was added at a dose of 100 µg/day.
Two days after total hypophysectomy, desmopressin was started because he showed polyuria, hypernatremia (serum Na, 157 mmol/L), and high serum osmolality (330 mosm/kg) without concentrated urine (urine osmolarity, 96 mosm/kg).
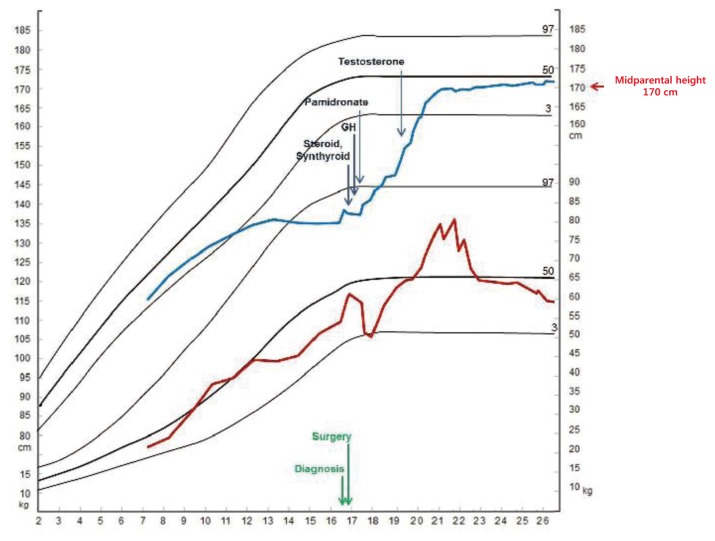
Within a month of the total hypophysectomy, recombinant human GH therapy was initiated at a dose of 0.02 mg/kg/day for 6 days a week subcutaneously. Two months after the total hypophysectomy, intravenous pamidronate was administered at a dose of 1 mg/kg/day over 4 hours for three days every 4 months for three years. Testosterone enanthate was initiated at a dose of 250 mg monthly at age 19 years.
The growth rate of the patient increased to 6 cm over the initial one-year follow-up. At age 21, recombinant human GH therapy was discontinued (Fig. 2). After another six months, the peak growth hormone level, as determined by insulin tolerance test, was 0.1 ng/mL, indicating adult GH deficiency. GH therapy subsequently was restarted at a dose of 0.4 mg/day. At his current age of 26, the patient's final height has attained the target range (172.2 cm, -0.20 SDS); weight (58.7 kg, -0.85 SDS). His BMD of femur neck and lumbar spine were 0.679 g/cm2 (z-score= -4.3 ) and 0.555 g/cm2 (z-score= -3.0), respectively.
At age 26 years, the patient's testis volume was 1 mL and sexual maturity rating of pubic hair is Tanner stage 4. He has been under treatment with testosterone enanthate, desmopressin acetate, prednisolone, levothyroxine sodium, and recombinant human GH for combined pituitary hormone deficiency and adult GH deficiency.
Discussion
This report described the long-term course of a Cushing disease patient with his growth profile, pubertal status, and responses to hormone replacement therapy, and emphasized the importance of optimizing growth, puberty, and bone health in patients who have undergone transsphenoidal surgery.
Selective adenomectomy by the transsphenoidal approach is the treatment of choice for Cushing disease; partial or total hypophysectomy is an alternative choice that carries a high risk of hypopituitarism or inadequate remission9). Nevertheless, surgical therapy, as compared with medical or radiation therapy, boasts a superior remission rate and a low risk of complications10).
When biochemical data on normal sellar MRI suggest Cushing disease, the remission rate varies from 50% to 70% according to surgical efficacy, and hypopituitarism and/or central diabetes insipidus often results. The recurrence rate after curative surgery is close to 25%11). Long-term surgical success can be predicted by the early-postoperative serum cortisol levels: levels below 2 µg/dL, for example, are associated with a low risk of recurrence12). Moreover, Patil et al.13) found a 2.5 times higher recurrence rate in patients with postoperative cortisol levels exceeding 2 µg/dL. Owing to persistently elevated serum cortisol and 24-hour urine cortisol levels, the patient in the present case, with pituitary microadenoma showing right lateralization of ACTH secretion by BIPSS, underwent total hypophysectomy two weeks after the initial right hemihypophysectomy. Other treatment options for recurrent or persistent disease after transsphenoidal surgery include bilateral adrenalectomy, conventional radiotherapy, or medical therapy using ketoconazole or metyrapone, which inhibits steroidogenesis14,15).
Long-term exposure to hypercortisolemia in untreated Cushing disease brings about significant morbidity, such as malignant hypertension, cardiovascular disease, hyperglycemia, osteoporosis, psychiatric features such as depression or cognitive dysfunction, and even mortality10,16,17). Our patient had osteoporosis at the time of diagnosis, and was treated with pamidronate for three years. After this course of therapy, his BMD was restored to the normal range. After his total hypophysectomy, combined pituitary hormone deficiency was documented by combined anterior pituitary function test, and subsequently, hormone replacement therapy with recombinant human GH, levothyroxine, glucocorticoid, and oral desmopressin were administered.
In conclusion, the long-term clinical course of a patient with Cushing disease who underwent transsphenoidal surgery was reported. For such patients, lifelong monitoring is needed in order to manage subsequent diverse endocrinological complications such as GH deficiency, hypogonadotropic hypogonadism, corticotropin deficiency, central hypothyroidism, and osteoporosis.