Response to three years of growth hormone therapy in girls with Turner syndrome
Article information
Abstract
Purpose
Short stature is the most common finding in patients with Turner syndrome. Improving the final adult height in these patients is a challenge both for the patients and physicians. We investigated the clinical response of patients to growth hormone treatment for height improvement over the period of three years.
Methods
Review of medical records from 27 patients with Turner syndrome treated with recombinant human growth hormone for more than 3 years was done. Differences in the changes of height standard deviation scores according to karyotype were measured and factors influencing the height changes were analyzed.
Results
The response to recombinant human growth hormone was an increase in the height of the subjects to a mean value of 1.1 standard deviation for subjects with Turner syndrome at the end of the 3-year treatment. The height increment in the first year was highest. The height standard deviation score in the third year was negatively correlated with the age at the beginning of the recombinant human growth hormone treatment. Different karyotypes in subjects did not seem to affect the height changes.
Conclusion
Early growth hormone administration in subjects with Turner syndrome is helpful to improve height response to the treatment.
Introduction
Turner syndrome (TS) is a disorder with distinct clinical features resulting from the complete or partial absence of one of the X chromosomes in females. The incidence is estimated to occur in 1 out of 2,500 live births1). The manifestations may vary according to the range of chromosomal aberration. However, hypergonadotropic hypogonadism occurs in virtually all affected individuals and short stature is a major confronting issue among patients. The mean adult height in untreated patients is approximately 20 cm shorter than that of the general female population2,3).
Impaired response to growth hormone (GH) is thought to be the major cause of retarded growth in TS subjects rather than GH deficiency4). Although recombinant human growth hormone (hGH) has been prescribed to improve adult height in TS patients for decades5-7), only a few case-control studies have addressed the effect of hGH administration on the individual and the optimal age for commencement of hGH has not been established yet. Recent studies have suggested that initiating hGH treatment at a younger age is more effective8-10).
This study is a retrospective analysis of patients with TS who were treated with hGH for more than three years. We investigated factors that were associated with the height response to the treatment.
Materials and methods
1. Subjects
Clinical records of twenty-seven girls with TS were reviewed. Patients selected were diagnosed with TS between 2003 and 2008 based on peripheral blood karyotyping, treated with hGH for at least 3 years, and were followed at the pediatric endocrinology unit of a tertiary hospital located in metropolitan Korea. Patients with chronic systemic illness, hypothyroidism, or a history of prior treatment with hGH were excluded. The dose of hGH was 1.0 IU/kg/wk, given six times per week (0.3 mg/kg/wk).
2. Methods
The subjects visited the outpatient clinic for follow-up every 3 months and their height, weight, complete blood cell counts, routine chemistry, and thyroid function were monitored. Height was measured using a Harpenden stadiometer marked to the nearest 0.1 cm and weight was recorded to the nearest 0.1 kg with an electric scale. Bone age was assessed every 6 months and interpreted by one investigator according to the Greulich and Pyle method11). Midparental height (MPH) was calculated as the mean of parental height minus 6.5 cm. Height standard deviation scores (SDS) were used to evaluate the first, second and third year responses to hGH treatment. The response to hGH was assessed using the height SDS in the third year of treatment and its changes from each of the previous years. The SDS were calculated according to the 2007 growth reference of Korean children and adolescents by Korean Pediatric Society and Korea Centers for Disease Control and Prevention and the reference growth chart for Korean girls with TS12).
The Institutional Review Board of the hospital approved this study and informed consent was obtained from all parents and patients.
3. Statistics
The continuous variables were expressed as mean±standard deviation for normally distributed variables. The Kruskal-Wallis test was used to evaluate differences among karyotypes. Annually measured parameters associated with height changes were assessed with paired T tests. Multiple regression analysis was used to evaluate the relationship between the height outcome and independent factors of clinical parameters. Data were analyzed using SPSS ver. 15.0 (SPSS Inc., Chicago, IL, USA). P-values<0.05 were used as the cutoff for statistical significance.
Results
1. Auxological outcome according to karyotype
Table 1 shows karyotypes of subjects with TS, participated in this study. Auxological characteristics according to karyptypes are summarized in Table 2. Twenty-seven patients were treated with hGH for more than 3 years and no adverse events such as type 2 diabetes, slipped capital epiphyses, or leukemia were observed in the subjects during the treatment period. The initial age was 8.6±2.8 years and 45, monosomy X karyotype was found in 22.2%, followed by 46, X, i(Xq). The MPH SDS was -0.3±0.5 and was slightly below the mean height. The initial height SDS for the general population was -2.7±0.8 and 95% of the subjects were below the 7th percentile. That was 0.3 standard deviation (SD) above the mean for the growth chart of Korean girls with TS. The Kruskal-Wallis test demonstrated no significant differences in initial age, bone age, height and weight and their SDS in between the karyotypes.
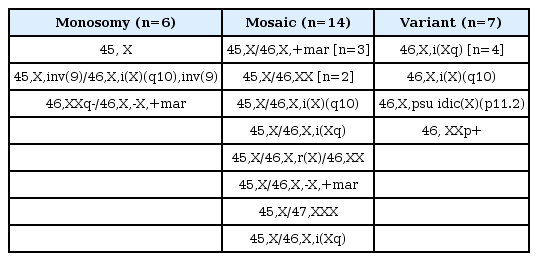
Karyotypes of 27 patients with Turner syndrome
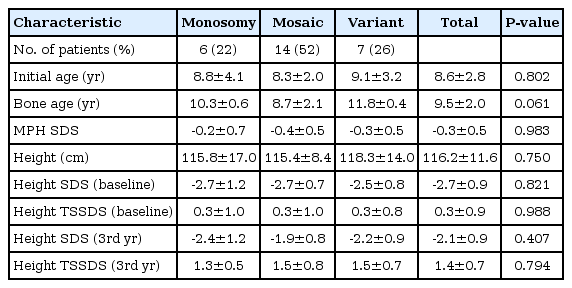
Baseline characteristics and clinical profiles according to karyotype
2. Height response during treatment period
The administration of hGH was observed to increase the heights of the subjects to a mean value of 1.1 SD (95% confidence interval, 0.9 to 1.3; data not shown) for the TS growth chart and a mean value of 0.58 SD (95% confidence interval, 0.3 to 0.8; data not shown) for the normal population by the end of the third year. The height increment in the first year was the highest and statistically significant, and the effect in the second year also showed a significant raise in the height standard deviation score for Turner syndrome (TSSDS) and height SDS (Fig. 1). The trend in height gain continued in the third year, although no statistical significance was observed (P=0.346) for the normal population. Although subjects with mosaic karyoptypes tended to have a slightly higher height TSSDS, no statistically significant difference was observed in height gain according to the karyotypes (Fig. 2). The height SDS in the third year was negatively correlated with the age at the initiation of hGH treatment (Fig. 3). The age at commencement of treatment was inversely correlated with height SDS at the third year in the multiple regression analysis adjusted for confounders, such as the difference between bone age and chronological age at the start of treatment, and MPH SDS (Table 3).
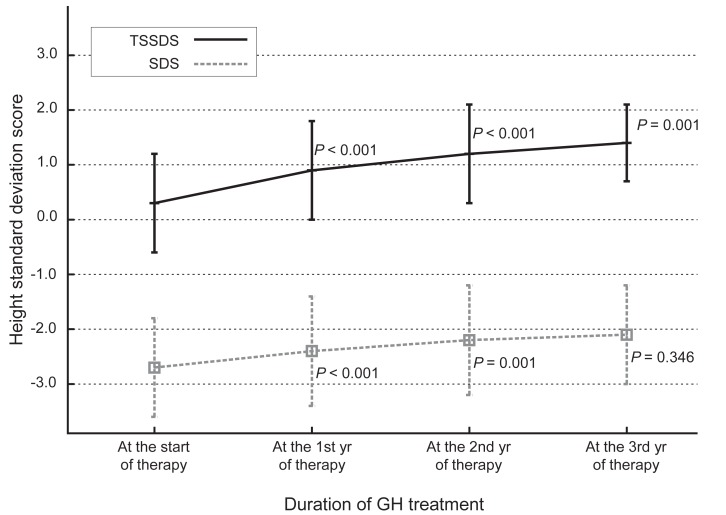
Changes in height standard deviation scores for Korean Turner syndrome (TSSDS, continuous line) and normal population (standard deviation score [SDS], dashes) patients during growth hormone (GH) treatment. P-values indicate the differences between each point in time and the previous observation.
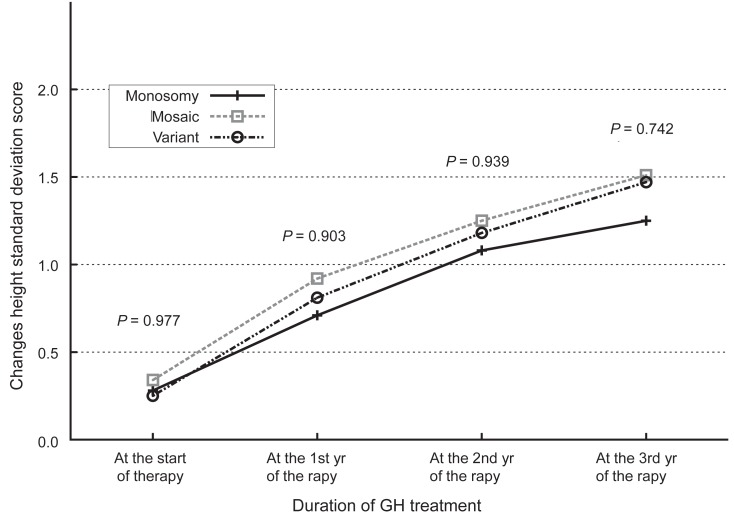
Changes in standard deviation score for the height of Korean Turner syndrome patients according to the karyotypes of the subjects. GH, growth hormone.
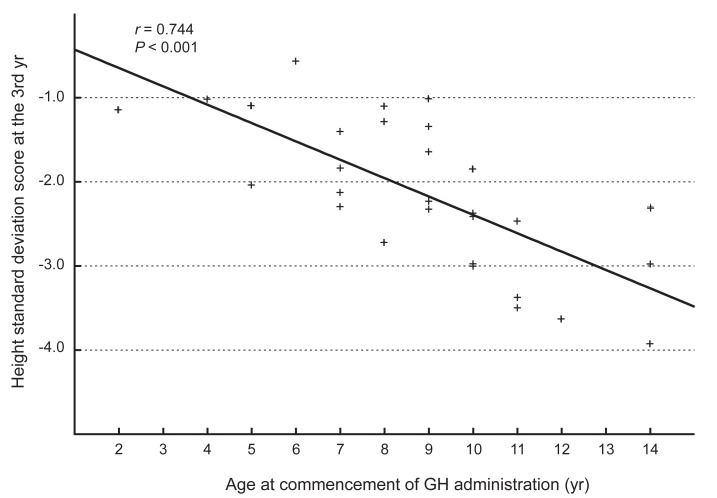
Relationship between the age at the commencementof growth hormone (GH) administration and the standard deviation scores of height at the third year.
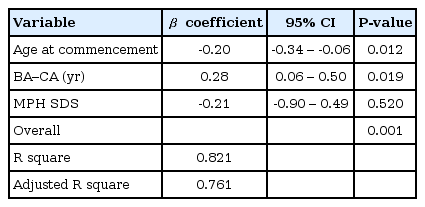
Multiple regression analysis of the third year height SDS
Discussion
This study was performed to identify the effect of hGH on subjects with TS and to find clinical factors that may be associated with the outcome of the hGH administration. The height gain was greatest in the first year of treatment and it was negatively associated with the age of commencement. The differences in the subjects'karyotypes did not affect the outcome of hGH administration or the height gain. Although subjects with variant karyotypes showed a slight tendency towards more accretion in height SDS, this was not statistically significant. This finding is compatible with previous reports13-15).
It has been reported that the age at commencement of hGH therapy and initial height SDS were negatively correlated with height gain, while MPH SDS had positive correlation with the growth outcome13,14,16). Sas et al.17) demonstrated that high doses of hGH improved adult height in TS patients. In this study, the age at commencement of hGH treatment and height SDS at the third year showed the negative correlation during the treatment period.
Short stature is the most common clinical presentation in girls with TS. The correction of short stature is reported as beneficial for the patient's quality of life18) and psychological adjustment may be achieved through the height gain, resulting in improved social competence. Girls with TS show growth failure during early childhood. The height gain in affected girls deteriorates during puberty and they usually do not have the normal pubertal growth spurt. The etiology of growth failure in TS is not well understood. Some studies19-21) reported that GH secretion decreased in subjects with TS whereas normal GH secretion was observed by other authors22,23). It is not clear whether the retarded growth is a result of resistance to GH or GH deficiency. It is supposed that haploinsufficiency of the short stature homeobox (SHOX) gene contributesmore to the short stature than a GH-IGF axis disturbance24, 25).The SHOX gene, which is located on the distal end of the X chromosome at Xp22.3 in the pseudoautosomal region 1, is known to act as a repressor of growth plate fusion. Although short stature and premature ovarian failure are virtually universal findings in patients with TS, recent studies suggest that the distal short arm of the X chromosome are mainly responsible for growth in affected girls, and deletion of proximal region of the X chromosome contributes to the deterioration of ovarian function26,27).
The additional height improvement in TS patients with hGH administration varies between 1 cm28) to 10 cm29,30). Chung et al.31) reported that 29 Korean patients with TS who were treated with hGH were 4.1 cm taller than who did not. Shin et al.32) observed 3.4 cm increment of final height in 121 Korean patients with TS. As with heightgain after hGH administration in patients with idiopathic GH deficiency, subjects with TS appear to be most efficacious during the first year of treatment33). Bakker et al.34) observed that younger subjects in the first year of hGH treatment demonstrated better results in their growth. The height gain in the subjects of this study was also greatest in the first year of treatment and more profound in younger patients. The height of the patients who started the treatment before 12 years of age increased 5.6 to 12.3 cm whereas those who received treatment after 12 years of age only gained 4.5 to 5.8 cm in height during the first year.
Davenport et al.35) demonstrated that hGH administration for patients with TS aged between 9 months and 4 years was followed by attained height SDS within the normal range, whereas the height SDS of nontreated control patients continuously decreased. Linglart et al.36) also reported patients receiving treatment before 4 years of age could achieve height gain without significant adverse effects. Early intervention for height improvement has a number of benefits in girls with TS, such as improvement in their relationships with peers, better self-image, and estrogen replacement at an appropriate age. While an earlier study37) reported that growth of girls with TS is preserved during their preschool age, it is now accepted that growth failure begins even in the first few years of life38,39).
In this study, the proportion of subjects with monosomy X was much lower than the known prevalence because this study was not a population-based trial. A recent study reported that the incidence of adverse events with hGH administration, such as increased intracranial pressure or slipped capital femoral epiphysis was higher in subjects with TS than in those for other indications40). Though no adverse events with the hGH administration were reported during the treatment period, this study is retrospective in design so loss to follow-up can be assumed.
In conclusion, early treatment of hGH in patients with TS seems to be essential for significant height gain and careful monitoring should accompany hGH treatment in patients with TS.
Notes
No potential conflict of interest relevant to this article was reported.