Various endocrine disorders in children with t(13;14)(q10;q10) Robertsonian translocation
Article information
Abstract
Purpose
45,XY,t(13;14)(q10;q10) karyotype can suggest infertility associated with more or less severe oligospermia in male adults. In addition, 45,XX,t(13;14)(q10;q10) karyotype carries reproductive risks such as miscarriage or infertility in female adults. However, reports on the phenotype of this karyotype in children are very rare. This study was done to observe various phenotypes of this karyotype in children.
Methods
Between January 2007 and December 2012, children diagnosed with 45,XY,t(13;14)(q10;q10) or 45,XX,t(13;14)(q10;q10) karyotype by chromosome analysis were analyzed retrospectively.
Results
Eight children (5 boys and 3 girls) were diagnosed with 45,XY,t(13;14)(q10;q10) or 45,XX,t(13;14)(q10;q10) karyotype. They ranged in age from 5 years and 6 months to 12 years and 4 months. The phenotypes of the study patients consisted of 1 hypogonadotrophic hypogonadism, 1 precocious puberty, 3 early puberty, 2 growth hormone deficiency (GHD) (partial) and 1 idiopathic short stature. As shown here t(13;14)(q10;q10) Robertsonian translocation shows a wide range of phenotypes.
Conclusion
It can be said that t(13;14)(q10;q10) Robertsonian translocation shows various phenotypes from GHD to precocious puberty in children. Further large-scale studies are necessary.
Introduction
Robertsonian translocations involve 2 acrocentric chromosomes (chromosomes 13, 14, 15, 21 and 22) that occur in 1/1000 births1). Translocation (13;14) is one of the most frequent form with an approximate 75% among Robertsonian translocations1,2).
Carriers of Robertsonian translocations have normal phenotype, but can have problem of infertility associated with more or less severe oligospermia in male adults3-5). In addition, 45,XX,t(13;14)(q10;q10) karyotype carrys reproductive risks such as miscarriage or infertility in female adults6). However, reports on phenotype of this karyotype in children are very rare. This study was done to see various phenotypes of this karyotype in children.
Materials and methods
Between January 2007 and December 2012, children who were diagnosed with 45,XY,t (13;14)(q10;q10) (Fig. 1A) or 45,XX,t(13;14)(q10;q10) (Fig. 1B) karyotype by chromosome analysis were analyzed retrospectively. For children with precocious puberty or early puberty or hypogonadotrophic hypogonadism, chromosome analysis was performed as a screening test to find out if they have any unknown chromosomal anomalies. Girls with short stature underwent chromosome to rule out variant Turner syndrome. A chromosomal study was also performed if one of parents has t(13q;14q). For chromosome analysis, peripheral blood lymphocytes were cultured. And then, karyotyping by the GTG banding (G-bands trypsin Giemsa) metaphase chromosome analysis was performed.

(A) Karyotype of a 12-year-old boy with hypogonadotrophic hypogonadism. (B) Karyotype of a 8-year-old girl with precocious puberty. Arrows indicate abnormal chromosomes.
Results
Eight children were enrolled. Five boys presented with 45,XY, t(13;14)(q10;q10) karyotype and their average age was 10.3±2.0 years (6.9 to 12.3 years). Three girls had 45,XX,t(13;14)(q10;q10) karyotype and their average age was 7.2±1.5 years (5.5 to 8.2 years). Chromosome analysis was recommended to all of the family members but 6 parents refused the test (cases A, B and F). All eight children and their parents showed normal morphology with no organ anomalies. Pedigree charts of all families were drawn (Fig. 2).

Pedigree chart of all families. Solid symbols represent affected individuals; open symbols, unaffected individuals; +, individuals with t(13;14)(q10;q10); -, individual with negative chromosome study. Only members assigned letters A through H were evaluated. A, a boy with hypogonadotrophic hypogonadism; B, a girl with growth hormone deficiency (partial); C-III, a boy with early puberty; D-III, a girl with central precocious puberty; E-III, a boy with early puberty; F, a girl with early puberty; G-III, a boy with growth hormone deficiency(partial); H, a boy with idiopathic short stature.
Case A was a 12-year-old boy presented for evaluation of cryptorchidism. He was diagnosed as cryptorchidism (right) at the age of 4-year-old but had not been treated. His father was 176 cm tall and his mother was 158 cm tall. His mother had a history of two miscarriages. His height was 150.9 cm (50th percentile). His weight was 45.2 kg (50th percentile). On genital examination, his right testis could not be palpated. His left testis was 1 mL in volume and descended into his scrotum. The stretched penile length was 3.5 cm (less than 2.5 standard deviations below the mean for his age). His pubic hair was Tanner stage 1. The diagnosis of hypogonadotrophic hypogonadism was made by low lutenizing hormone (LH), follicle-stimulating hormone (FSH), and testosterone. His level of LH was <0.1 IU/L (normal, 0.2 to 4.9 IU/L) and FSH was 0.21 IU/L (normal, 1.8 to 3.2 IU/L). His testosterone measured <0.2 ng/mL (normal, 0.18 to 1.5 ng/mL). The level of inhibin B was 14 pg/mL (normal, 41 to 328 pg/mL). Blood samples were obtained 0, 15, 30, 45 and 60 minutes after stimulation with 100 mcg intravenous gonadotropin-releasing hormone (GnRH) agonist (gonadorelin, Relefact LH-RH) for examination of FSH, LH and total testosterone levels. FSH (0.21, 1.03, 1.78, 2.17, and 2.67 IU/L, respectively), LH (<0.1, 0.864, 1.3, 1.22, and 1.15 IU/L, respectively) and extremely low testosterone levels was detected(<0.2 ng/mL, respectively). The baseline serum concentrations of thyroid-stimulating hormone (1.44 µIU/mL; normal range, 0.7 to 6.4 µIU/mL) and prolactin (4.96 ng/mL; normal range, 3 to 18 ng/mL) were normal. His bone age was 11 years by the standards of Greulich and Pyle at chronologic age 12 years and 4 months. Testicular ultrasound of the right inguinal canal reveals an undescended testis of 1.1 cm. No anatomical abnormalities were detected in brain magnetic resonance imaging (MRI) (Fig. 3A). It is difficult to differentiate between hypogonadotrophic hypogonadism and constitutional delay of puberty. But diagnosis of this case is hypogonadotrophic hypogonadism first, especially considering his chromosome abnormality that may cause infertility associated with hypogonadotrophic hypogonadism. In addition, his parents didn't have histories of short stature in children, delayed puberty, and eventual normal stature.

(A) Brain magnetic resonance imaging (MRI) of case A with hypogonodotrophic hypogonadism. No anatomical abnormalities are detected in brain MRI. (B) Brain MRI of case B with growth hormone deficiency. MRI of the hypothalamic pituitary region is normal. (C) Brain MRI of case G with growth hormone deficiency. Brain MRI doesn't detect any abnormality.
Case B was a girl visited our clinic for evaluation of short stature at age 5 years and 6 months. On examination, the patient measured 104.1 cm (less than 3th percentile) and weighed 16 kg (3th percentile). Although physical examination was unremarkable except short stature, a chromosome study was performed to rule out variant Turner syndrome and diagnosed with 45,XX,t(13;14)(q10;q10). Her father was 167 cm and her mother was 150 cm tall. Her bone age was 3 years and 10 months by the standards of Greulich and Pyle. Initial laboratory studies revealed the following: Insulin-like growth factor 1 (IGF-1) 164 ng/mL (between -1 standard deviation [146 ng/mL] and mean [242 ng/mL] value), normal thyroid function and normal adrenal function. The evaluation of growth hormone (GH) secretion showed a partial GH deficiency (GHD; GH peak response to L-dopa, 9.4 ng/mL; GH peak response to insulin, 9.4 ng/mL). Magnetic resonance imaging of the hypothalamic-pituitary region was normal (Fig. 3B).
Case C was a 11-year-old boy had the karyotype 45,XY, t(13;14)(q10;q10) and presented with early puberty. His father was 172 cm tall and had normal kayotype. His mother was 155 cm tall and had 45,XX,t(13;14)(q10;q10) karyotype. General physical examination of the patient was within normal limit. On genital examination, his testis was 15 mL in volume his pubic hair was Tanner stage 1. His height was 153.5 cm (90th percentile), his weight 39.8 kg (50th percentile). In this patient bone age (13 years and 3 months) was greater than chronologic age (11 years and 4 months). Hormonal laboratory tests, including serum prolactin (10.9 ng/mL; normal range, 3 to 18 ng/mL), dehydroepiandrosterone sulfate (DHEA-S; 153.7 ug/dL, normal range, 48 to 200 µg/dL), thyroid stimulating hormone (2.53 µIU/mL; normal range, 0.5 to 4.8 µIU/mL), free thyroxine (1.39 ng/dL; normal range, 0.8 to 2.3 ng/dL), testosterone (2.71 ng/mL; normal for age, 1.0 to 3.2 ng/mL), FSH (1.45 IU/L; normal range, 1.2 to 5.8 IU/L), and LH (1.52 IU/L; normal range, 0.2 to 5.0 IU/L) were all within normal range for age. Peak LH in response to GnRH stimulation test was 13.7 IU/L, and peak FSH 1.49 IU/L. Brain magnetic resonance imaging did not detect any abnormality in his brain or pituitary gland.
Case D was a 8-year-old girl had the karyotype 45,XX,t(13;14) (q10;q10) and presented with precocious puberty. Her father was 177 cm tall and had normal kayotype. Her mother was 154 cm tall and had 45,XX,t(13;14)(q10;q10) karyotype. This patient visited our pediatric endocrinology unit for evaluation of bilateral breast budding. On examination, her breast enlargement was compatible with Tanner stage 2 and pubic hair Tanner stage 1. Her height was 121.3 cm (25th percentile), weight 23.5 kg (25th to 50th percentile). Her bone age was 7 years and 10 months at chronologic age 8 years. A hormonal assay revealed normal levels of estradiol 6.94 pg/mL (normal, 5 to 20 pg/mL); prolactin, 12.4 ng/mL (normal, 3 to 24 ng/mL); DHEA-S, 45.68 µg/dL (normal, 19 to 114 µg/dL); FSH, 3.56 IU/L (normal, 1.0 to 4.2 IU/L); and LH, <0.1 IU/L (normal, 0.02 to 0.18 IU/L). Her stimulated level of LH was 3.61 IU/L and stimulated FSH 15.1 IU/L.
Case E patient was referred to our department at the age of 11 years, because of accelerated growth recently and was diagnosed with early puberty. His father was 170 cm tall and had 45,XY,t(13;14)(q10;q10). His mother was 160 cm tall and had normal karyotype. His mother had a miscarriage once. A physical examination revealed a Tanner stage 2-3 for penis and pubic hair development. His testicle volume was 15 mL. The patient measured 144 cm (50th to 75th percentile), weighed 55.9 kg (90th to 95th percentile) and BMI 26.96 kg/m2 (greater than 97th percentile). His bone age was 12 years and 6 months. GnRH stimulation test revealed pubertal response of gonadotropins (peak LH, 15.8 IU/L). MRI of the brain was normal.
Case F was a 8-year-old girl had the karyotype 45,XX,t(13;14) (q10;q10) with early puberty. This patient visited our pediatric endocrinology unit for evaluation of unilateral breast budding. Her father was 180 cm tall and her mother was 161 cm tall. No significant family history noted. On examination, her right breast enlargement was compatible with Tanner stage 1-2 and pubic hair Tanner stage 1. Anthropometric examination revealed following; height was 130.8 cm (75th percentile), weight 27.9 kg (50th to 75th percentile). Her bone age was 9 years and 4 months at chronologic age 8 years and 3 months. Thyroid profile done was normal. Hormonal assay revealed following; FSH, 22.3 IU/L (normal, 1.0 to 4.2 IU/L); LH, 16.2 IU/L (normal, 0.02 to 0.18 IU/L); prolactin, 22.1 ng/mL (normal, 3 to 24 ng/mL); estradiol, 9.46 pg/mL (normal, 5 to 20 pg/mL); 17-hydroxyprogesterone, 1.78 ng/mL (normal, 0.15 to 2.21 ng/mL). Her stimulated level of LH was 3.31 IU/L and stimulated FSH 18.2 IU/L. Brain MRI was normal. Pelvis ultrasonography didn't reveal any demonstrable pathology.
Case G patient underwent chromosome study at the age of 4 years since his father had been diagnosed with 45,XY,t(13;14) (q10;q10). His mother had normal karyotype and had a miscarriage. Both of parents got chromosome analysis to find out the reason of the previous miscarriage. His father was 179 cm and mother was 155 cm tall. On examination, the patient measured 131.2 cm (10th to 25th percentile) and weighed 35.5 kg (50th percentile). He was investigated for his height at the age of 10 years and 1 month with a bone age of 5 years and 9 months. IGF-1 level was 122.3 ng/mL (between -2 standard deviation [67 ng/mL] and -1.5 standard deviation [126 ng/mL] value). Peak levels of GH were 5.39 ng/mL after L-dopa administration and 7.72 ng/mL after insulin administration respectively. He was diagnosed with GHD (partial). MRI of the hypothalamic pituitary region was normal (Fig. 3C).
Case H, this boy is a younger brother of case G. He underwent chromosome study at the same time with case G for the same reason and was proven to have the karyotype 45,XY,t(13;14) (q10;q10). His height was 112.2 cm (below 3th percentile) and weight was 18.6 kg (below 3th percentile). His bone age was 3 years at chronologic age 6 years and 11 months. Initial laboratory studies revealed normal thyroid function and normal adrenal function. IGF-1 level was 151.0 ng/mL (between -1.5 standard deviation [113 ng/mL] and -1 standard deviation [153 ng/mL] value). GH peak response to L-dopa was 8.37 ng/mL and GH peak response to insulin was 24.69 ng/mL on the GH stimulation test. He was diagnosed with idiopathic short stature (ISS) because he had normal levels of GH in response to provocative tests with significant bone age delay. It was also considered the fact that his parents didn't have histories of constitutional delay of growth and puberty to make the diagnosis.
All children's information is summarized in Table 1. In our study, we have patients consist of 1 hypogonadotrophic hypogonadism, 1 central precocious puberty, 3 early puberty, 2 GHD (partial) and 1 ISS.
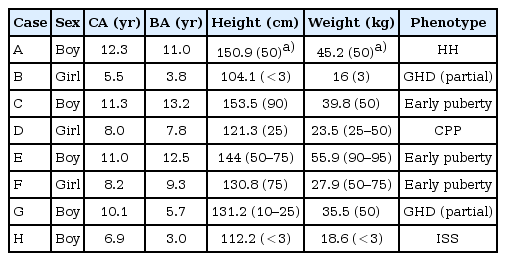
Summary of patients
Discussion
Robertsonian translocation involving chromosomes 13 and 14 occur with a prevalence of 0.97 in 1,000 in general population, which is the most frequent Robertsonian translocations7). This structural chromosome abnormality is associated with infertile men3-5). In addition, literature shows women with this karyotype can have spontaneous abortions6,8).
Luciani et al.9) investigated pachytene analysis of a man with t(13q;14q) and they reported the interaction between X chromosome in the sex vesicle, which is formed in male pachytene and t(13q;14q) interferes with X chromosome inactivation. This interaction may cause male infertility such as oligospermia or azoospermia and its rate was 61%9). This rate explains the reason why the rest carriers of male have no problem with fertility.
It is well known that the translocation carrier's pregnant possibility is directly associated with the number of balanced or normal spermatozoa. Pellestor et al.10) reported the proportion of normal and balanced gamatesamong spermatozoa from t(13q;14q) carriers was 74%. Other two studies showed the rate of normal and balanced gamate was 94% and 87% respectively11,12). Escudero et al.13) analyzed chromosome in sperm from two 45,XY,t(13;14)(q10;q10) carriers. His study showed the frequency of balanced spermatozoa was 74% and 77% in each patient.
Morel et al.14) reported there was an increased incidence of chromosome 8 disomy in the spermatozoa of the patient with 45,XY,t(13;14)(q10;q10) karyotype, whose son has a mosaic chromosome 8 trisomy. Moreover, there was also a significant increase in chromosome 18, 21, XX and YY disomies in the spermatozoa of the patient.
Careful reproductive risk assessment and genetic counselling will be provided for children with this kayotype through regular follow-up.
In our study, children with 45,XY,t(13;14)(q10;q10) or 45,XX,t(13;14)(q10;q10) karyotype showed various endocrine disorder such as hypogonadotrophic hypogonadism, GHD (partial) and ISS. For children with precocious puberty or early puberty, chromosome analysis was performed as a screening test to find out if they have any unknown chromosomal anomalies. We finally found 4 patients with t(13;14)(q10;q10) Robertsonian translocation among 168 children consisted of precocious puberty or early puberty between January 2007 and December 2012. Therefore, chromosome analysis needs to be considered for children with precocious puberty or early puberty.
Cases C, D, E and F had brain MRI for our study to detect any anatomical abnormalities in brain. But MRI of their brain was normal.
Further large-scaled studies are necessary to find out the interaction between various endocrine phenotypes and the Robertsonian translocation involving chromosomes 13 and 14.
Notes
No potential conflict of interest relevant to this article was reported.