The first case of hyperosmolar diabetic ketoacidosis in a patient diagnosed with MODY 5 (maturity-onset diabetes of the young type 5) and 17q12 microdeletion syndrome
Article information

Highlights
· Here, we report the case of a 12-year-old girl diagnosed with maturity-onset diabetes of the young type 5 (MODY 5) who presented with hyperosmolar diabetic ketoacidosis followed by acute complications such as thrombocytopenia-associated multiple organ failure, cerebral microbleeds, and diabetic amyotrophy.
To the editor,
Hyperosmolar diabetic ketoacidosis (HODKA) is a severe acute complication of diabetes mellitus (DM) and the leading cause of death in children and adolescents with DM.
An 11-year-old girl with a loss of consciousness visited the emergency room. She had shown typical hyperglycemic symptoms (polydipsia, polyuria, hyperphagia, and 16% weight loss) for 20 days. Her father had history of type 2 DM. The patient was born as a term baby of 3,200 g through vaginal delivery without perinatal problems.
On admission, her height was 140.0 cm (4.7th percentile), and her weight was 30.0 kg (2.4th percentile). She had a semicomatose mentality (Glasgow Coma Scale 6), hemodynamic shock, and an early stage of severe coccygeal sore and ecchymosis on both legs. Her plasma glucose level was 501 mg/dL, glycosylated hemoglobin 15.4%, serum osmolality 336 mOsm/kg, Na/K/Cl 139/2.4/120 mmol/L, blood urea nitrogen/creatinine 47.6/1.88, Ca/P/Mg 7.1/7.4/1.5 mg/dL, aspartate aminotransferase/alanine aminotransferase 146/41 IU/L, creatine kinase (CK) 8,478 U/L (reference, 0–190 U/L), CK muscle brain 101.3 ng/mL (reference, 0–3.6 ng/mL), high sensitivity troponin I 3,986.94 pg/mL (reference, 0–11.6 pg/mL), myoglobin >3,909.0 ng/mL (reference, 12–80 ng/mL), brain natriuretic peptide 1,845 pg/mL (reference, 0–100 pg/mL), prothrombin time/activated partial thromboplastin time 16.2/48.6 seconds (reference, 12.7–16.1/33.9–46.1 seconds), fibrinogen 267 mg/dL (reference, 200–400 mg/dL), AT III activity 39% (reference, 80–120), D dimer 2.95 μg/mL (reference, <0.5 μg/mL), fibrin degradation product 11.82 μg/mL (reference, <5 μg/mL), and platelets 65,000/μL. An arterial blood gas analysis showed pH 6.86, pCO2 66 mmHg, bicarbonate 11.8 mM, and base excess -20.7. In the subsequent test results, plasma c-peptide was 0.442 ng/mL, 24-hour urine c-peptide was 2.264 μg/day, anti-glutamic acid decarboxylase antibody was 83.62 U/mL (reference, <1.00 U/mL), and urine glucose/ketone/protein was 2+/-/2+. a disintegrin and metalloproteinase with thrombospondin type 1 motif 13 (ADAMTS-13) activity was 48% (reference, 50%–1,160%), and von Willebrand factor (vWF) antigen was 182.2% (reference, 52.2%–177.9%).
She was initially diagnosed with type 1 DM and HODKA. She required mechanical ventilation and had complications of thrombocytopenia-associated multiple organ failure (TAMOF), rhabdomyolysis, and coccygeal sores.
She could not stand alone, and she complained of severe pain in her left hip with passive range of motion. Computed tomography and venography of the lower extremities showed no evidence of a thrombus. However, the left quadriceps femoris muscle had a decreased volume without nerve compression. A nerve conduction velocity test and electromyography showed polyneuropathy and myopathies in the bilateral lower extremities, more severe on the left than the right, compatible with diabetic amyotrophy.
On the 21st hospital day, her body weight was profoundly decreased (6 kg over 4 days) even though her blood sugar level was well controlled. Brain magnetic resonance angiography revealed brain atrophy and multifocal microbleeds at the corpus callosum, left frontoparietal lobe, left internal capsule, both thalami, right basal ganglia, and pons (Fig. 1). A chromosomal microarray test was performed due to intellectual disability (IQ 47), and a 17q12 microdeletion (1.3 Mb) was found, including the HNF1B gene (Fig. 2).
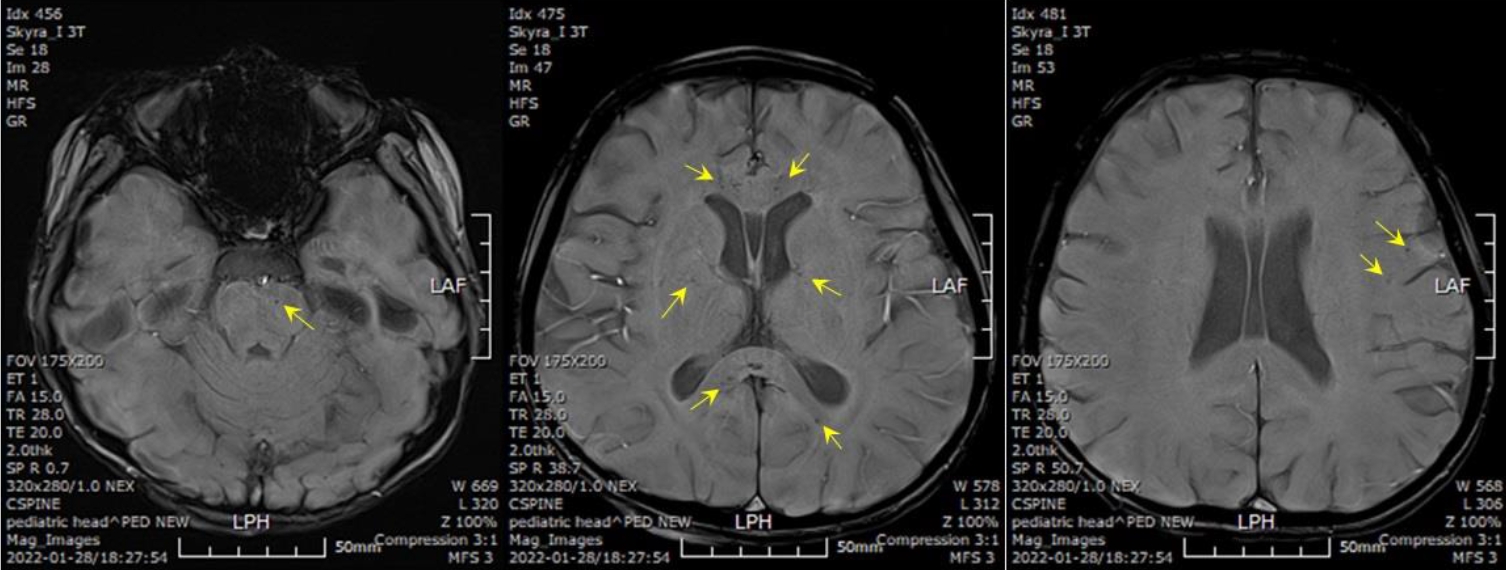
Brain magnetic resonance angiography reveals multifocal microbleeds at the corpus callosum, left frontoparietal lobe, left internal capsule, both thalami, right basal ganglia, and pons, as indicated by the yellow arrows.
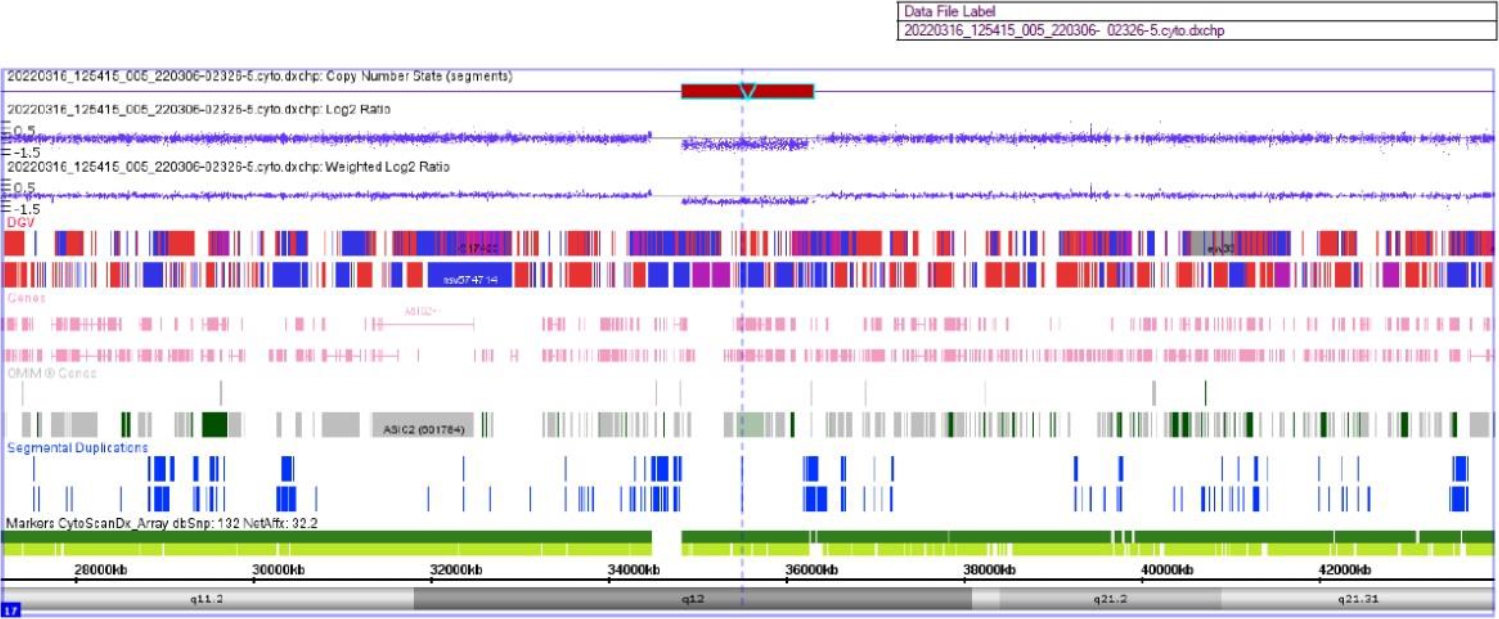
Chromosomal microarray test (CytoScan Dx assay, Genome build hg19) results show approximately 1.48 Mbp of microdeletion at 17q12 (34,822, 465-36, 307, 773). The 17q12 microdeletion in this area contains HNF1B, the causative gene of MODY 5 (maturity-onset diabetes of the young type 5).
TAMOF is characterized by thrombocytopenia with progressive multiple organ failure in critically ill patients. Thrombocytopenia reflects its involvement in disseminated microvascular thromboses [1]. Therefore, in a hyperglycemic emergency situation, early detection and proper management of TAMOF are very important to decrease the risk of related morbidity, and the platelet count should be monitored frequently [2].
Diabetic ketoacidosis (DKA) can cause endothelial damage, and vWF mediates platelet adhesion to damaged endothelial tissues [2]. After that, ADAMTS-13 has a role in cleaving vWF. However, TAMOF involves an ADAMTS-13 deficiency. The mechanism of ADAMTS-13 is unclear, but ADAMTS-13 gene mutations and inhibiting antibodies were revealed in thrombotic thrombocytopenic purpura [1]. Noncleaved vWFs cause multiple microthromboses and increase the risk of TAMOF [2]. It may be helpful to predict rapid progression to TAMOF by measuring ADAMTS-13 level at the initial time when thrombocytopenia is detected.
Meanwhile, the pathophysiology of diabetic amyotrophy seems to be ischemic nerve fiber degeneration and vasculitis because of poor tissue perfusion [3]. It causes motor dysfunction and neuropathic pain, along with sudden weight loss. The prognosis is good for most people, but some have residual motor dysfunction and require a wheelchair [3]. Therefore, it is necessary to evaluate motor function and body weight after the severe hyperglycemic crisis has passed.
To our knowledge, this is the first report of a HODKA presentation in a pediatric patient with MODY 5 and 17q12 microdeletion syndrome. Identifying hyperosmolarity in HODKA is important because its treatment requires more aggressive fluid administration than does in DKA, and improper management can increase the risk of complications.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics statement
This report was approved by the Institutional Review Board (IRB) of Pusan National University Yangsan Hospital, Yangsan, Korea (IRB No. 05-2023-001). Written informed consent for publication of this case report was obtained from the patient's parent.