Resistance to thyroid hormone and nonfunctioning pituitary microadenoma in a 13-year-old boy with THRB mutation
Article information

Highlights
· We describe a case of a boy diagnosed with resistance to thyroid hormone (RTH) who also had a nonfunctioning pituitary microadenoma. This is the first report of a pediatric case of coexistent RTH and a nonfunctioning pituitary microadenoma.
To the editor,
Resistance to thyroid hormone (RTH) is a rare syndrome characterized by elevated serum free iodothyronine and nonsuppressed thyroid stimulating hormone (TSH) levels. The most common cause of RTH is heterozygous pathogenic mutations in the thyroid hormone receptor β (THRB) gene. The incidence of RTHβ is estimated to be 1:40,000–1:50,000, and about 85% of RTHβ cases have a THRB gene mutation [1,2]. Although both RTHβ and TSH-secreting pituitary adenoma (TSHoma), which has a biochemical profile similar to RTH, are rare, accurate differential diagnosis is necessary to ensure appropriate treatment. Patients with RTHβ do not require treatment, while patients with TSHoma require surgical removal. We herein present the case of a boy diagnosed with RTHβ who also had a nonfunctioning pituitary microadenoma.
A 13-year-old Korean boy presented to the hospital with palpitations, insomnia, and headache. Laboratory tests revealed elevated serum T3 and free T4 (fT4) levels and TSH in the normal range. At that time, he was diagnosed with hyperthyroidism and prescribed an antithyroid drug (methimazole, 20 mg/day). Increasing the methimazole dose due to persistently elevated fT4 caused further aggravation of the patient's palpitations, insomnia, headache, and goiter and new onset of general weakness, mild dyspnea, and chest discomfort. Pituitary magnetic resonance imaging (MRI) revealed a 3-mm delayed enhancing nodular lesion in the right portion of the pituitary gland with suspicious minimal stalk deviation, which was suspected to be a pituitary microadenoma. At age 14 years, he visited our hospital for surgical treatment for a pituitary microadenoma. He still complained of palpitations, insomnia, general weakness, and mild dyspnea with chest discomfort, and he had a grade I goiter. Laboratory investigation revealed elevated serum T3 and fT4 levels and inappropriately elevated TSH (serum T3 246 ng/dL, TSH 19.3 μIU/mL, fT4 3.38 ng/dL). Thyroid technetium scintigraphy showed diffuse enlargement with increased radioactive technetium uptake. Thyroid ultrasound showed diffuse thyroid enlargement and multiple small colloid cysts. The effective tests for differentiating RTHβ and TSHoma are serum level of glycoprotein hormone α-subunit (α-GSU) and the α-GSU/TSH molar ratio, which are elevated in TSHoma and within the normal range in RTHβ. Additionally, for most patients with TSHoma, TSH does not increase in thyrotropin-releasing hormone (TRH) stimulation or T3 suppression tests, whereas RTHβ patients show a normal TSH response. Neither his α-GSU level (0.45 μg/L; reference range, 0.05–0.66 μg/L) nor his α-GSU/TSH molar ratio were elevated (0.23, normal <1), and his TRH stimulation test showed a normal TSH response, which was compatible with an RTH diagnosis. At that point, his pituitary lesion was thought to be a nonfunctioning pituitary microadenoma, not TSHoma. Currently, genetic testing for the THRB gene is the gold standard for RTHβ diagnosis. Whole-exome sequencing revealed a heterozygous mutation, c.1021C>G (p.Leu341Val), in THRB (Fig. 1), which was previously reported in TRH [3]. This is a likely pathogenic variant according to the Human Gene Mutation Database (HGMD professional version 2022 [4]. The patient's mother and sister had the same mutation, and they both showed the same thyroid hormone profile pattern, although they did not have any symptoms related to hyperthyroidism. The patient's father and older brother did not have the mutation and had normal thyroid hormone profiles (Fig. 1). Thus, methimazole treatment was stopped, and his clinical symptoms, including the mild dyspnea with chest discomfort and general weakness, resolved. Also, the palpitations and insomnia were ameliorated by treatment with a β-adrenergic blocking agent. The patient's TSH level decreased to within the normal range after methimazole was stopped. The serial changes in thyroid hormone levels and clinical symptoms are shown in Fig. 2.
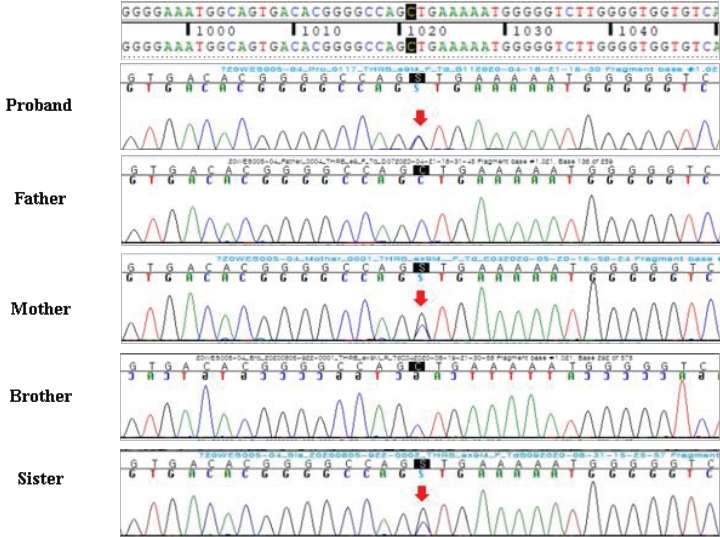
Chromatogram of the heterozygous mutation, c.1021C>G, (pLeu341Val), in the THRB gene of the proband, his mother, and elder sister. This mutation was not detected in the father or brother.
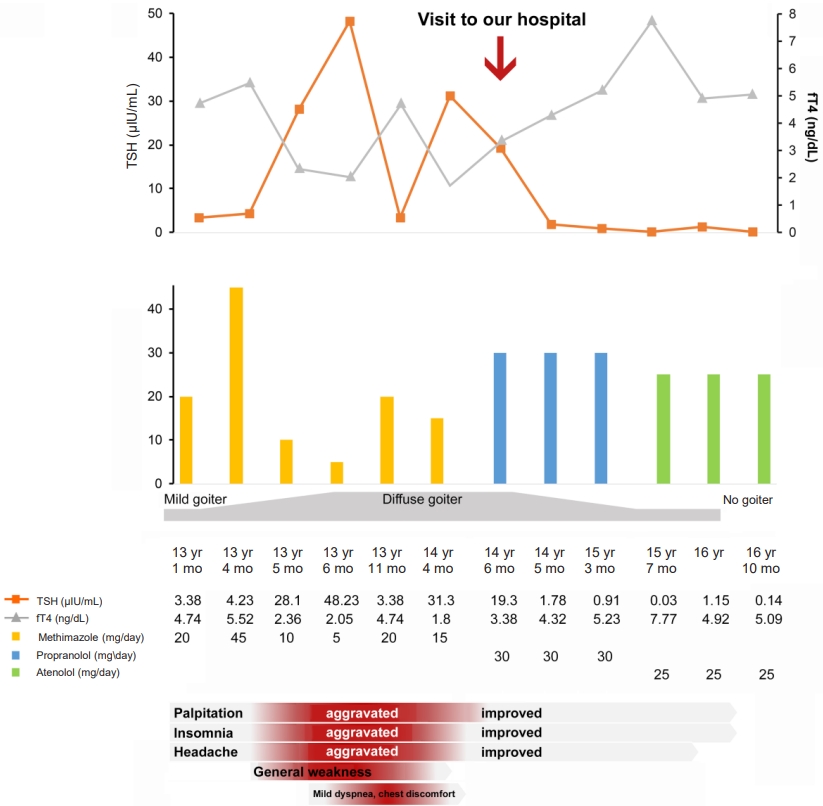
Serial changes in clinical symptoms with thyroid hormone and medication use. TSH, thyroid stimulating hormone; fT4, free T4.
When elevated serum-free T4 and T3 and normal or high serum TSH levels are present, TSHoma and RTH are suspected. Interestingly, this patient also had a pituitary microadenoma, detected by MRI, which raised the question of RTHβ or TSHoma. For an accurate diagnosis and to avoid inappropriate treatment, differential diagnosis is essential because these 2 diseases should be managed with completely different treatments. RTHβ is usually asymptomatic or variously nonspecific, and severity varies widely. Goiter is the most common sign, observed in 65%–95% of RTHβ patients, followed by tachycardia in 30%–75%. In addition, goiter can be accompanied by short stature; developmental delays; abnormal psychology; attention deficit disorder; and repeated infections of the ears, nose, and throat [1,4]. In most reported RTHβ cases, patients do not require treatment for increased thyroid hormone because of adequate compensation. Symptoms of thyrotoxicosis, such as palpitation or tremor, can be treated with a β-adrenergic blocking agent. Some thyroid hormone analogues, such as 3,3ʹ,5-triiodothyroacetic acid, 3,3ʹ,5,5ʹ- tetraiodothyroacetic acid, 3,5-diiodothyropropionic acid, and dextro(D)-T4, have been studied in RTH [5-8]. Our patient complained of palpitations, insomnia, and general weakness, and his palpitation and insomnia improved after he started taking a β-adrenergic blocking agent, despite persistently high serum fT4 level. His mother and sister had the same mutation and had high serum fT4 with normal TSH level, but they were asymptomatic and did not require treatment.
In conclusion, we report a 13-year-old boy with both RTHβ and a nonfunctional pituitary microadenoma. This case shows that faulty thyroid hormone secretion requires careful diagnosis using adequate biochemical, genetic, and radiographic approaches to prevent diagnostic delays and avoid unnecessary and potentially harmful disease management.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics statement
Informed consent was obtained from the parents of the patient.