Highlights
· This is a case of a 9-year-old Korean boy with diabetes caused by a known heterozygous mutation in NR0B2. He had a positive family history of diabetes, tested negative for autoantibodies, and showed normal pancreatic function at diagnosis. The patient had a likely pathogenic variant of c.293_301delinsAC (p.Leu98Hisfs*6) in NR0B2.
To the editor,
Maturity-onset diabetes of the young (MODY) is inherited in an autosomal dominant manner and is typically diagnosed before the age of 25 years. Since MODY was first reported by Tattersall [1] in 1974, several causative mutations of the disease have been identified. New genetic mutations causing MODY and their pathogenesis are under investigation, as it is thought that many causative genes remain to be identified [2].
NR0B2, first discovered in 1996, is an important modulator of signaling pathways related to cholesterol, bile acids, fatty acids, and glucose metabolism [3]. Studies have reported that mutations in this gene seem to be associated with diabetes and also appear to cause decreased insulin sensitivity [4-6]. However, mutations in NR0B2 have not been widely reported; thus, the clinical characteristics of patients with NR0B2 mutations and their relationship with diabetes and obesity remain unclear.
Herein, we report a case of diabetes with a known heterozygous mutation in NR0B2.
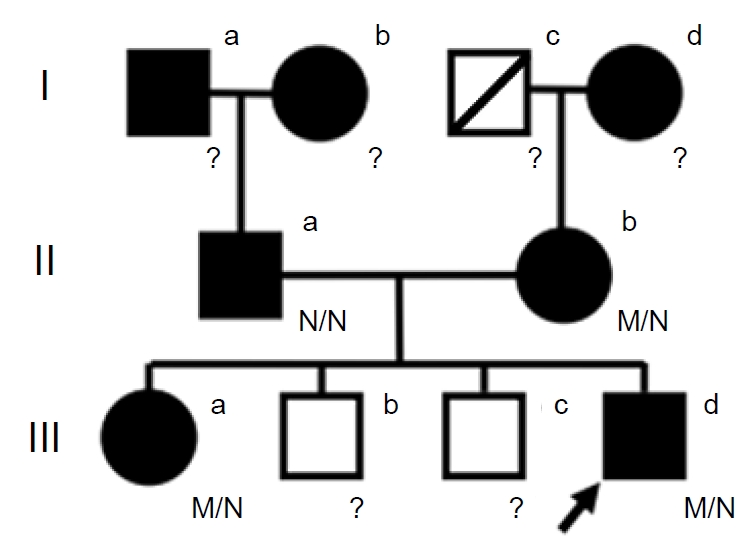
A 9-year-old boy presented with abdominal pain and vomiting for 2 days. He was delivered via cesarean section at a gestational age of 40 weeks and had a birth weight of 3.7 kg (61st percentile). His height was 137.5 cm (62nd percentile), his weight was 40.0 kg (85th percentile), and his body mass index was 21.2 kg/m2 (89th percentile). His grandparents and parents had type 2 diabetes, and his older sister had been diagnosed with type 1 diabetes at 10 years of age (Fig. 1).
His initial blood glucose level was 297 mg/dL; he had glucosuria but not acidosis. Glycated hemoglobin A (HbA1c) was 10.6%, and auto-antibodies (islet cell antibodies, antiglutamic acid decarboxylase antibodies, and insulin auto-antibodies) were undetectable. An oral glucose tolerance test showed a serum glucose level of 205 mg/dL in the fasting state and of 261 mg/dL after 2 hours. His serum insulin levels were 11.60 μIU/mL (fasting) and 14.80 μIU/mL (2 hours), and his serum C-peptide levels were 2.80 ng/mL (fasting) and 3.62 ng/mL (2 hours). Homeostasis model assessment of insulin resistance (HOMA-IR), HOMA-β, and QUICKI (quantitative insulin check index) scores were calculated to evaluate insulin resistance; the patient showed values of 5.9, 29.4%, and 0.2962, respectively, indicating mild insulin resistance. His total cholesterol concentration was 80 mg/dL, his triglyceride concentration was 67 mg/dL, his high-density lipoprotein (HDL) cholesterol concentration was 28 mg/dL, and his low-density lipoprotein cholesterol concentration was 38.6 mg/dL. The patient was started on multiple daily injections of insulin degludec and lispro. After treatment was initiated, his blood glucose was well-controlled at a total insulin dose of approximately 0.6 U/kg/day, and his HbA1c level was maintained at 5.4%–6.2%.
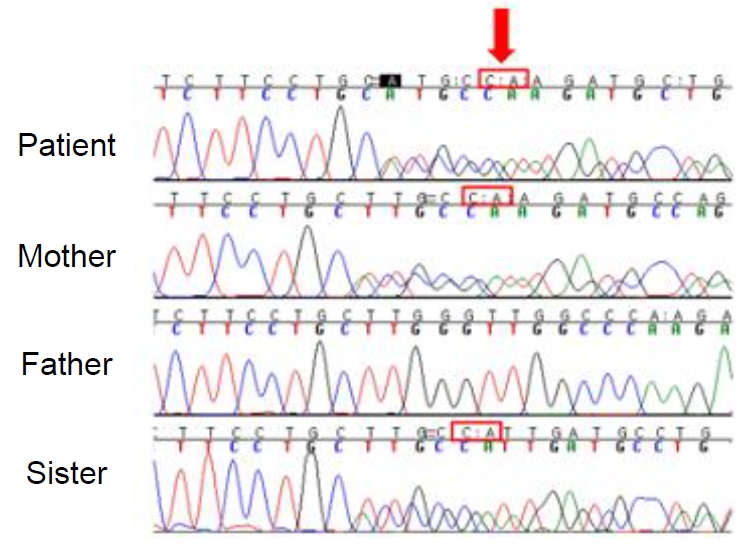
Next-generation sequencing revealed a heterozygous mutation (NM_021969.2: c.293_301delinsAC: p.Leu98Hisfs*6) in NR0B2, which was classified as likely pathogenic according to the American College of Medical Genetics and Genomics guidelines [7]. His parents and sister had undergone sequencing of the mutation site, and his mother and sister showed the same heterozygous mutation (Fig. 2).
NR0B2, also known as the small heterodimer partner (SHP), is a unique and atypical nuclear receptor. NR0B2, located on chromosome 1p36.1, spans approximately 2 kbs and contains 2 exons [6]. By acting on other nuclear receptors, it regulates several transcriptional activities, including fundamental biological and metabolic processes such as cholesterol, glucose, and bile acid metabolism; lipogenesis; steroid hormone biosynthesis; xenobiotic homeostasis/metabolism; and the cell cycle [3]. Particularly in the pancreas, this nuclear receptor represses the transcriptional activity of HNF1A and may disrupt the feedback circuit between HNF1A and HNF4A [5]. Therefore, mutations in NR0B2 may lead to diabetes with a pattern similar to that of MODY caused by HNF1A or HNF4A mutations [6].
Nishigori et al. [4] investigated the role of SHP in 173 unrelated Japanese subjects with early onset nonketotic diabetes. Six heterozygous mutations in SHP were identified in 7 subjects, revealing a clear association between SHP mutations and obesity. All subjects with these mutations were obese at the time of diabetes diagnosis; they also had higher birth weights and were more obese compared to their family members. Meanwhile, a study of a large United Kingdom Caucasian cohort, including 458 subjects with young-onset type 2 diabetes, was performed to determine the role of SHP mutations in diabetes, obesity, and birth weight, yet no associations were detected [5].
HNF4A regulates the expression of proteins associated with lipid metabolism. In particular, HNF4A regulates apolipoprotein transcription, and HDL-cholesterol is mainly composed of apolipoprotein AII; as such, loss of HNF4A function is associated with reduced circulating concentration of HDL-cholesterol [8]. Some patients with MODY type 1 have low levels of triglycerides, apolipoprotein AII, and apolipoprotein CIII [9]. The observed values in this case were similar in the patient and his sister. As mentioned above, NR0B2 may interfere in the feedback circuit between HNF1A and HNF4A, and mutations in NR0B2 can lead to diabetes with a similar pattern as that observed in MODY caused by HNF1A and HNF4A mutations. These factors may explain the relationship between HNF4A and NR0B2. Diabetes due to NR0B2 mutations may be similar to MODY caused by mutations in HNF1A or HNF4A and may be treated using the same methods, such as oral sulfonylurea.
This letter supports the hypothesis that mutations in NR0B2 cause MODY by engaging in a pathway related to HNF regulatory pathways. Determining the biochemical action of NR0B2 by identifying its protein ligands will help reveal the mechanisms of its various functions, including energy metabolism and homeostasis, providing individual tools for effectively treating related diseases, such as diabetes and metabolic syndromes.