Highlights
· This study discusses a Kleefstra syndrome patient with micropenis. A chromosomal microarray revealed a 424-kb deletion at chromosome 9q34.3, including EHMT1. Early testosterone treatment initiated at 4 months led to improved penile growth.
Introduction
Kleefstra syndrome (OMIM #610253) is characterized by low intelligence, childhood hypotonia, and distinct facial features. Its diagnosis is made in a proband with heterozygous deletion at chromosome 9q34.3 or heterozygous pathogenic variants in the euchromatin histone methyl transferase 1 (EHMT1) gene [1-3], which regulates transcription by histone methylation [2,4]. Relevant phenotypes are autism spectrum disorder, hearing and vision impairment, congenital heart defects, renal defects, obesity, and seizures. Additionally, genital anomalies, such as micropenis, cryptorchidism, and hypospadias, are present in 30%–40% of male Kleefstra syndrome patients [1-3].
However, there is little research on the etiology, endocrine laboratory lab tests, and treatment focused on genital abnormalities in Kleefstra syndrome, whose etiology remains unknown. Torga et al. [5] reported a Kleefstra syndrome patient with micropenis treated with intramuscular testosterone cypionate injections. A case series [6] of Kleefstra syndrome patients did not report endocrine evaluations for associated genital anomalies. Herein, we report a Korean case of Kleefstra syndrome confirmed by a 424-kb deletion at chromosome 9q34. Endocrinological investigations and treatment effects of intramuscular testosterone injections for micropenis are described.
Case report
A male patient born at 38+5 weeks of gestation via normal vaginal delivery was transferred to Kyungpook National University Children's Hospital for assessment and management of imperforate anus. The patient had no specific family history. Routine antenatal findings during the pregnancy were normal. The patient was appropriate-for-gestational-age with a birth weight of 2,770 g (z-score: -0.87), height of 50 cm (z-score: 0.31), and head circumference of 35 cm (z-score: 0.72). Physical examination revealed facial dysmorphisms, including hypertelorism and anteverted nares. Genital examination revealed micropenis with stretched penile length measured at 0.9 cm (normal range: 3.38–3.92 cm, -2.5 standard deviation for age: 2.5 cm [7]), palpable testicles bilaterally, and no hypospadias.
Chromosomal analysis was performed on suspicion of a sex-development disorder, showing 46, XY. The patient had a sepsis-like condition after admission. To rule out adrenal insufficiency caused by congenital adrenal hyperplasia, laboratory investigation was performed at day 7 of life and showed normal electrolyte, adrenocorticotropic hormone (ACTH), cortisol, 17-hydroxyprogesterone, 11-deoxycortisol, and dehydroepiandrosterone-sulfate levels. Endocrinological assessment at the age of 3 months revealed low luteinizing hormone (LH, 0.14 mIU/mL; reference range [mean±SD] [8], 15.5±11.8 mIU/mL), follicle-stimulating hormone (FSH, 2.59 mIU/mL; reference range, 6.3±2.6 mIU/mL), testosterone (<3 ng/dL; reference range, 95±53 ng/dL), and dihydrotestosterone (7 ng/dL; reference range, 12–85 ng/mL) levels. To assess the function of Leydig cells, human chorionic gonadotropin (hCG) stimulation test was performed at the age of 3 months; 1,000 units of hCG were administered daily for 3 days. Testosterone levels increased from <3 ng/dL at baseline to 230 ng/dL after the final dose of hCG. Thus, we concluded that there was adequate testosterone biosynthesis in the Leydig cells. Chromosomal microarray was performed by single nucleotide polymorphism array method (CytoScan Dx Assays, Genome build: hg19) on the day of birth due to multiple congenital anomalies, including imperforate anus and micropenis. A 424-kb heterozygous deletion at 9q34.3 (arr[hg19] 9q34.3 (140,234,315-140,659,055) x1) was identified, which led to a diagnosis of Kleefstra syndrome. Involved OMIM genes were NOXA1, NSMF, PNPLA7, MRPL41, DPH7, ZMYND19, and EHMT1.
Screening tests for other associated phenotypic presentations were conducted. Echocardiogram showed a secundum atrial septal defect (3–4 mm) and patent ductus arteriosus (1.3 mm). Cranial magnetic resonance imaging was acquired at the age of 2 months and showed no congenital brain parenchyma or pituitary gland anomaly. Furthermore, an automated auditory brainstem response test result was normal for both sides. Renal ultrasound was unremarkable at initial study.
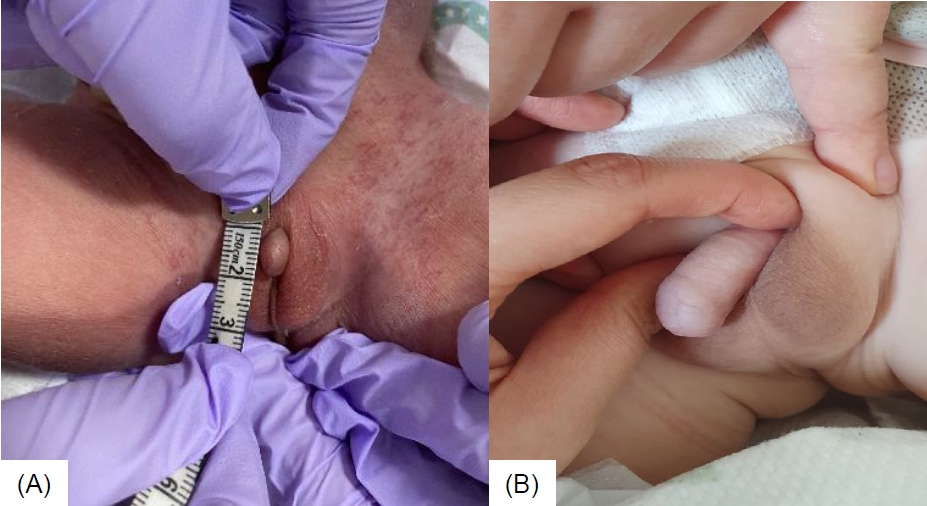
Intramuscular testosterone enanthate injections of 25 mg once every 3 weeks for 3 months was initiated at the age of 4 months. The patient showed a good response to testosterone injections with an improvement of stretched penile length from 0.9 cm to 3 cm (normal range, 3.5–5.1 cm; -2.5 standard deviation for age, 2.3 cm [7]) after 4 doses (Fig. 1A, B). After the fourth dose of testosterone injection, laboratory tests showed LH 0.04 mIU/mL, FSH <2.35 mIU/mL, and testosterone 220 ng/dL. There were no noted adverse reactions to the injections, such as hypertension, fluid retention, virilization, or polycythemia. The patient underwent double barrel colostomy 3 days postnatally, anoplasty at the age of 6 months, and colostomy repair at the age of 8 months. He had recurrent urinary tract infections, and grade 4 hydronephrosis in both kidneys developed at the age of 1 month. Additionally, grade 5 vesicoureteral reflux in the left kidney was noted, for which Deflux (hyaluronic acid/dextranome) injection was administered at the age of 12 months. Furthermore, the patient received physiotherapy for decreased muscle tone from the age of 6 months, and he walked at the age of 14 months. Bayley scales of infant and toddler development screening test conducted at the age of 15 months showed borderline delay of cognition and language, as well as motor delay. He received speech-language rehabilitation therapy from the age of 24 months. He had no seizure history.
The most recent physical examination at the age of 28 months showed height and weight of 93 cm (z-score: 1.32) and 14.8 kg (z-score: 1.43), respectively. The patient has received routine care in nephrology, pediatric colorectal surgery, urology, and rehabilitation medicine. He is being monitored at the endocrine clinic for growth velocity and pubertal progression.
This study was approved by the Institutional Review Board of Kyungpook National University Chilgok Hospital, Daegu, Korea (approval number: 2022-06-012). Informed case consent was obtained from the patient's parents or guardian for publication of this case report.
Discussion
We report the first Korean case of Kleefstra syndrome with micropenis confirmed by deletion in chromosome 9q34.3. We describe the progress of endocrine laboratory test results and treatment effect of testosterone therapy for micropenis.
The etiology of micropenis can be classified as hypergonadotropic hypogonadism (HH) due to primary testicular insufficiency, HH due to pituitary/hypothalamic insufficiency, defects in testosterone biosynthesis (such as 5-alpha reductase deficiency), developmental abnormalities, or idiopathic [7]. To determine the level of the hypothalamic-pituitary-gonadal axis affected, assessment of serum gonadotropins, pituitary hormones, testosterone, and its derivatives is necessary. Furthermore, testicular function should be evaluated with the hCG stimulation test.
Previous studies reported that micropenis, cryptorchidism, and hypospadias can accompany Kleefstra syndrome [1]. However, the etiology of these genital abnormalities remains unelucidated. In our patient, basal gonadotropins and testosterone levels were low at the age of 3 months, which is during the mini-puberty of infancy. The possibility of hypogonadotropic hypogonadism was not precisely elucidated because our patient was prepubertal and had not been tested for GnRH stimulation. Furthermore, Leydig cell function was considered normal because our patient showed adequate testosterone level increase in the hCG stimulation test.
Our patient had a 424-kb heterozygous deletion at 9q34.3 (arr[hg19] 9q34.3 (140,234,315-140,659,055)x1), confirmed by chromosomal microarray, and involved OMIM genes were NOXA1, NSMF, PNPLA7, MRPL41, DPH7, ZMYND19, and EHMT1. Kleefstra syndrome was diagnosed based on heterozygous deletion of the EHMT1. The characteristic facial features of Kleefstra syndrome and associated urogenital abnormalities were consistent with the genetic diagnosis. However, the etiology of genital anomalies in patients with EHMT1 pathogenic variants remains unclear. Recent studies [4,9] reported that various human diseases are associated with epigenetic dysregulation. Histone modification, especially methylation in H3, induces chromatin remodeling and controls transcriptional regulation [10,11]. EHMT1 methylates H3K9 in euchromatin, resulting in epigenetic transcriptional repression [4]. EHMT1 regulates transcription through methylation; furthermore, kisspeptin, a neuropeptide that plays a key role in gonadal development [12], also regulates GnRH neuron expression through histone acetylation/methylation [13,14]. Thus, a mutation in EHMT1 could repress transcription of the GnRH gene through histone methylation defect and epigenetic dysregulation.
Genital anomalies such as micropenis may be accompanied by a pathogenic variant in EHMT1 by itself, but mutation in the NSMF (OMIM #608137) may also be related to genital anomalies. NSMF is known to contribute to the HH phenotype in an oligogenic pattern, but monoallelic deleterious NSMF variants are not sufficient to cause HH [15,16]. Therefore, we considered that EHMT1 contributed to the genital anomaly in our patient.
Patients with micropenis have been shown to respond well to testosterone treatment for penile growth in infancy; therefore, it is important to initiate this treatment in early infancy [7,17]. Intramuscular injections of testosterone esters, such as testosterone enanthate or cypionate in oil, are long-acting agents, and 4 doses of 25 mg once every 3 weeks for 3 months should be used for initial treatment [7]. Our patient was treated with intramuscular testosterone enanthate injection at the age of 4 months, which led to an increase in penile size.
Previous reports [2,3] summarized the common features and growth profiles of Kleefstra syndrome patients: 25% had short stature and 45% were overweight. Torga et al. [5] reported a Kleefstra syndrome patient with micropenis (stretched penile length was 3 cm; normal range, 5.3–7.3 cm; -2.5 standard deviation for age, 3.8 cm [7]). He was treated with intramuscular testosterone cypionate injection at the age of 9 years 9 months. Follow-up exam at the age of 11 years showed stretched penile length of 6 cm (normal range, 5.3–7.5 cm; -2.5 standard deviation for age, 3.7 cm [7]). Our patient is not short or obese (height z-score, 1.32; weight z-score, 1.43), and improvement in penile length with testosterone treatment was observed (from 0.9 cm to 3 cm). The patient showed decreased muscle tone and speech delay, for which he has been receiving physiotherapy since the age of 6 months and speech-language rehabilitation therapy since the age of 24 months. Patients treated with sex hormone therapy should be monitored for pubertal progression. It is uncertain whether our patient will experience puberty, and evaluation of the hypothalamic-pituitary-gonadal axis with a GnRH stimulation test will be needed when the patient reaches pubertal age.
In conclusion, we report a Kleefstra syndrome case confirmed by deletion at chromosome 9q34.3; systematic endocrine work up should be performed in cases of combined genital abnormalities. Intramuscular testosterone injection treatment for micropenis was effective in our patient during early infancy. Further study via molecular biology techniques to determine the association between Kleefstra syndrome and GnRH neuron expression should be conducted to understand the etiology of genital anomalies in such patients.